Downloaded 980 times




































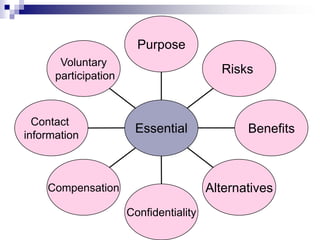





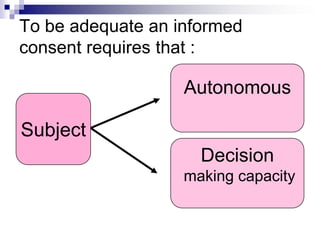





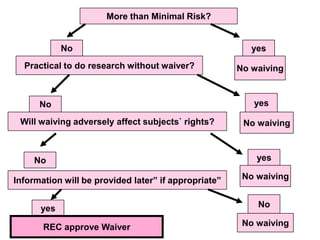




























This document outlines the key elements of obtaining informed consent, including what informed consent is, the ethical basis for requiring it, the consent process, what is needed for valid informed consent, elements that must be included, and special situations like children and waivers. It discusses how informed consent applies the ethical principle of respect for persons and facilitates trust in research. The consent process and what must be included in the consent form are described. Challenges like illiteracy, culture, child assent, and stored samples are also addressed.























![Indian gcp guidelines[647]](https://cdn.slidesharecdn.com/ss_thumbnails/indiangcpguidelines647-210325044800-thumbnail.jpg?width=640&height=640&fit=bounds)

















































