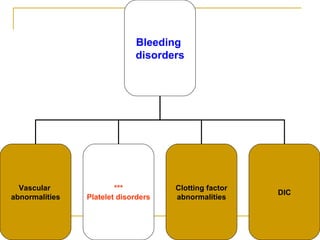
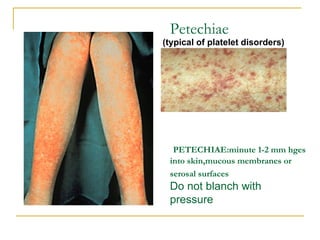
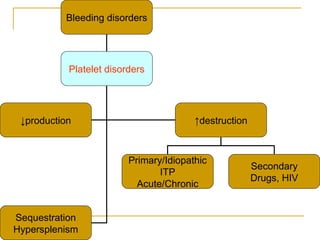
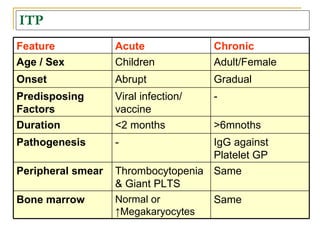
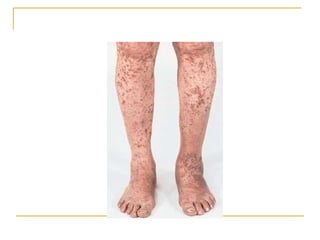
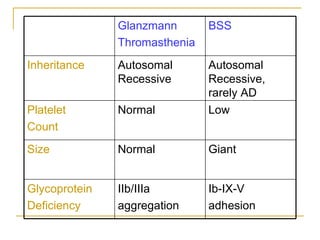
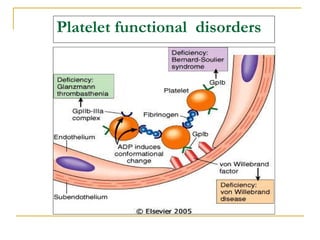
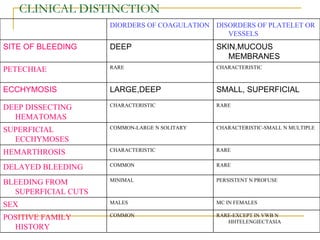
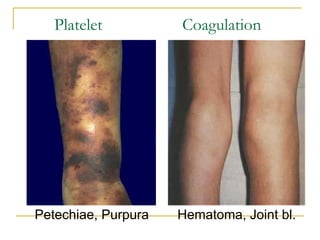
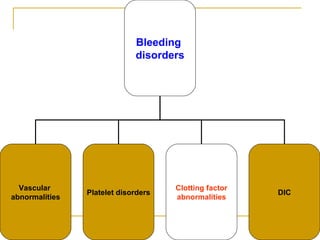
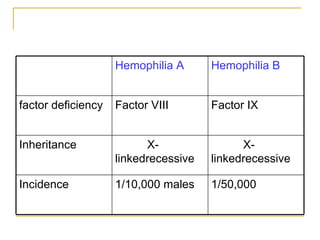
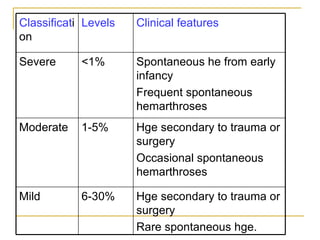
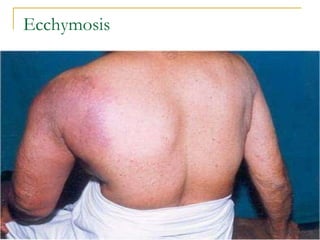
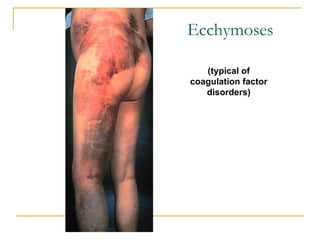
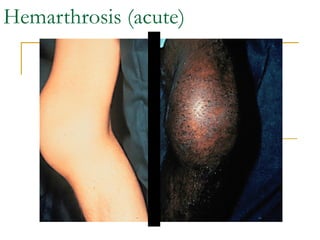
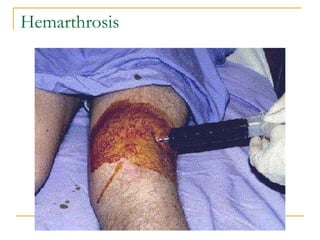
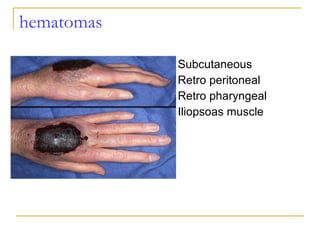
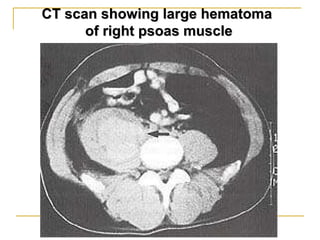
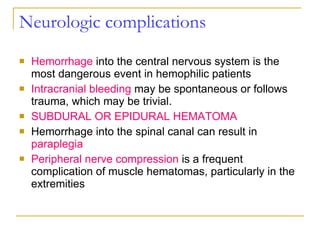
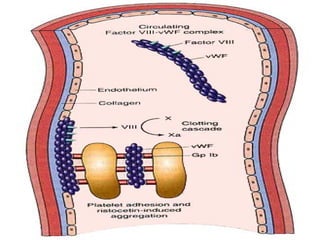
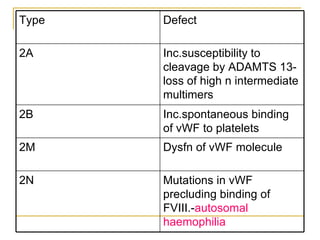
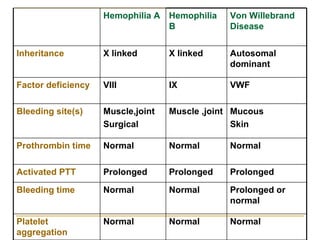
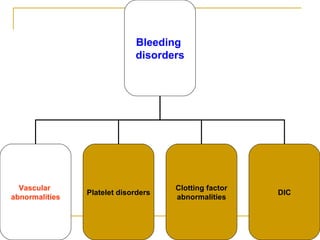
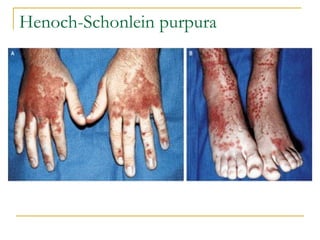
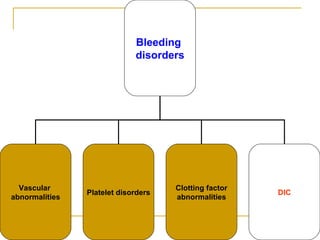
The document summarizes various bleeding disorders including their clinical features and classifications. Platelet disorders can cause petechiae and purpurae due to decreased platelet production, increased destruction, or sequestration. Clotting factor deficiencies like hemophilia A and B are inherited and cause hemarthroses, hematomas, and bleeding after trauma. Von Willebrand disease is the most common inherited bleeding disorder caused by a defect in von Willebrand factor.



















































































































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)
