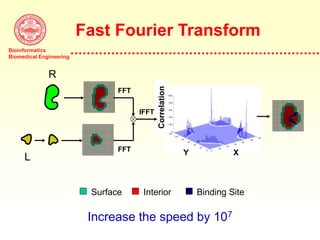
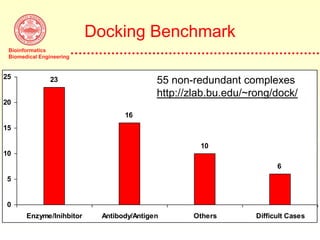
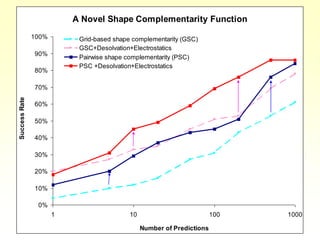
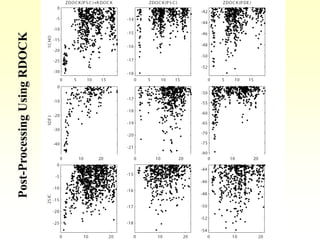
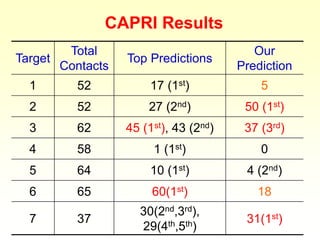
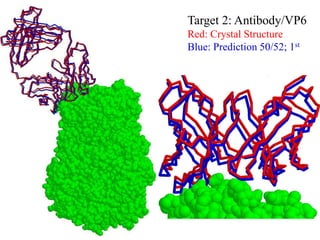
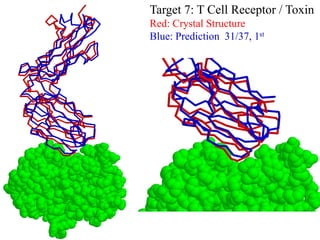
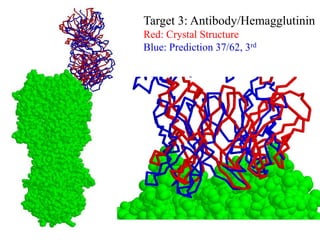
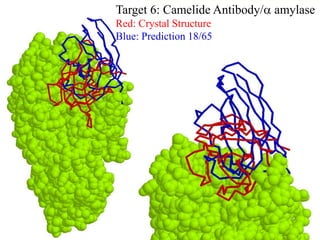
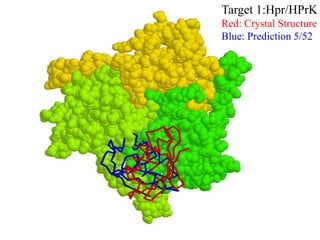
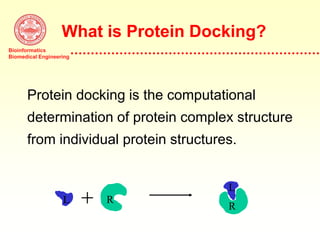
This document discusses protein docking, which is the computational determination of protein complex structures from individual protein structures. It describes challenges in protein docking like large search spaces and protein flexibility. The document outlines an integrated approach to protein docking that uses shape complementarity functions, desolvation energies, electrostatic interactions, and post-processing techniques. Evaluation on benchmark datasets shows a 60% success rate when 10 predictions are submitted. Future work proposed includes developing an automatic docking server and improving post-processing methods.





![Bioinformatics
Biomedical Engineering
Protein-Protein Interaction
Thermodynamics
Energy
Free
Binding
:
G
R
L
R
L
L
L
R
R
L
L
R R
L L
]
][
[
]
[
ln
L
R
RL
RT
G
water](https://image.slidesharecdn.com/zdockteaching-230319031732-d1d64a33/85/zdockTeaching-ppt-7-320.jpg)