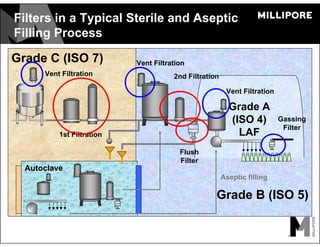
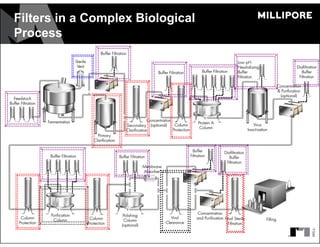
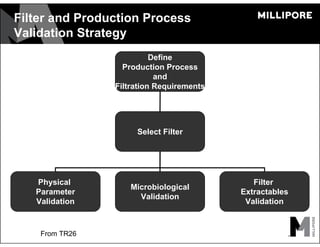
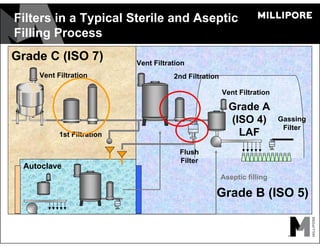
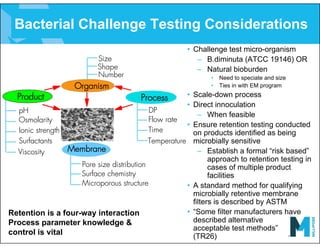
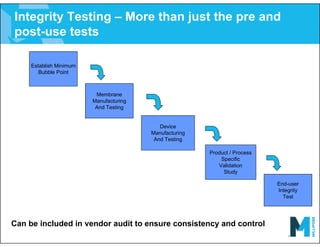
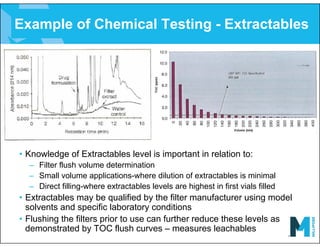
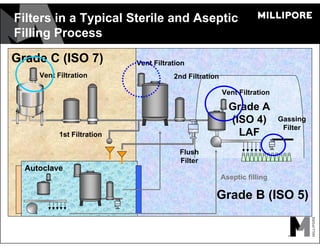
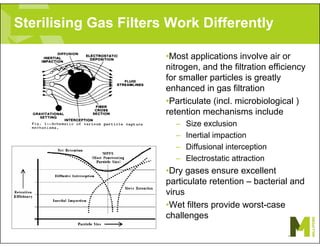
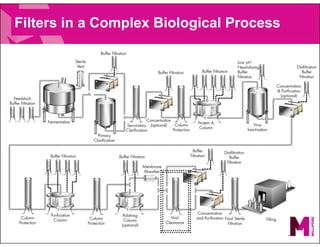
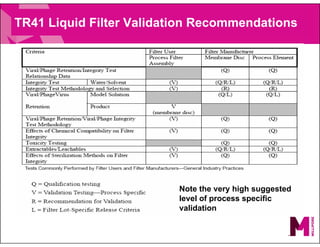
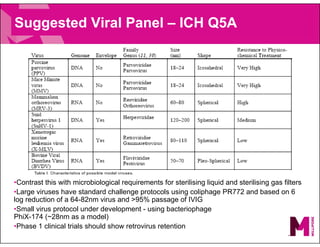
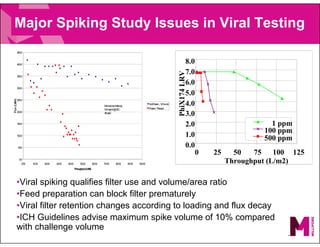
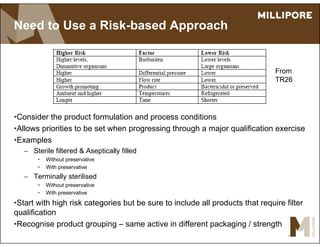
The document provides an overview of filter validation recommendations for sterilizing liquid filters, sterilizing gas filters, and virus filtration. It discusses key definitions, regulatory views, and recommendations from PDA technical reports. The presentation emphasizes using a risk-based approach and step-wise validation process. It recommends starting with higher risk categories and product groupings, and focusing validation efforts on filters used in critical applications.














































![Back To Basics GMPs[1]](https://cdn.slidesharecdn.com/ss_thumbnails/a9b29f4b-1869-4392-8cbd-d144ef2e7c57-160216213515-thumbnail.jpg?width=640&height=640&fit=bounds)


































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)


