This document provides a review of the Generic Drug User Fee Act (GDUFA) implemented by the US Food and Drug Administration (USFDA). It discusses:
1) The increasing backlog of generic drug applications and inspections that GDUFA aims to address by providing additional funds to the USFDA.
2) The key goals of GDUFA are to ensure safety, efficacy, and access by prioritizing application review, increasing facility inspections, and expediting approval processes.
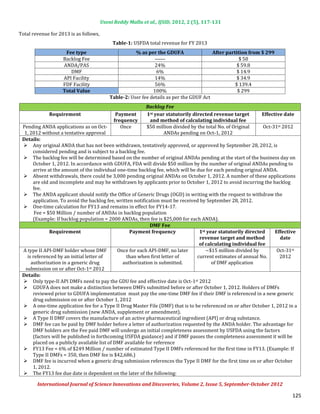
3) GDUFA fees are paid by generic drug manufacturers and facility owners and are split between application fees and facility fees, with the total revenue expected to be $299 million annually from 2013-




![Useni Reddy Mallu et al., IJSID, 2012, 2 (5), 117-131
Application Fees:
Backlog fees are in year 1 [(Year 1 Oct 1, 2012 to Sept 30, 2013) for ANDAs pending review at the date of program
Backlog fees:
implementation] and ANDA and Post-Approval Study (PAS) fees, as well as DMF first reference fees in all years.
Both finished dosage form manufacturer and API facilities with a modest fee differential reflecting the added costs of
Facility Fees:
overseas inspection.
Fees will be derived from both applications and facilities in a 30%-70% split. Fees will be split between finished
Source of Fees:
dosage form manufacturers and active pharmaceutical ingredient manufacturers in an 80%-20% split.
As per the GDUFA agreement all API, Finished Dosage Forms (FDF) facilities and applicants of DMF, ANDA are need to
GDUFA Fee agreement:
pay the user fee. Details of GDUFA fee agreement are,
Funding level = inflation adjusted $299M/year
Fees for Applications and Facilities
Applications in the backlog (year 1 only) Involved in manufacture of generic drugs,
Applicants Facilities
Drug master file fee whether Active Pharmaceutical Ingredient (API)
ANDA and prior approval supplement (PAS) filing or Finished Dosage Form (FDF), domestic or
fee foreign.
Exemption from fees: Positron Emission Tomography (PET) drugs
Individual fees calculated/published upon implementation and Order of magnitude lower than PDUFA fees
80% from finished dosage form manufacturers, 20% from API manufacturers
Critical splits:
70% from facility fees, 30% from application fees
In year 1, $50M from backlog fee, so above splits are slightly different
Figure-9: GDUFA user fee partition
International Journal of Science Innovations and Discoveries, Volume 2, Issue 5, September-October 2012
124](https://image.slidesharecdn.com/usfda-genericdruguserfeeactacompletereview-121228063313-phpapp02/85/Usfda-generic-drug-user-fee-act-a-complete-review-8-320.jpg)





















![cmc [ chemistry manufacturing control ]](https://cdn.slidesharecdn.com/ss_thumbnails/presentation2222ra-181120122336-thumbnail.jpg?width=640&height=640&fit=bounds)














































































