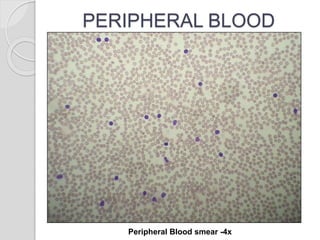
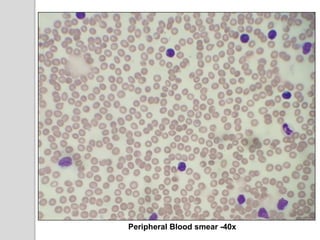
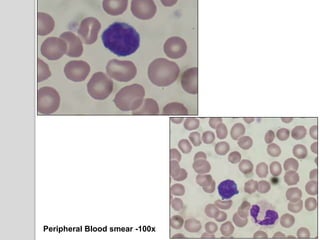
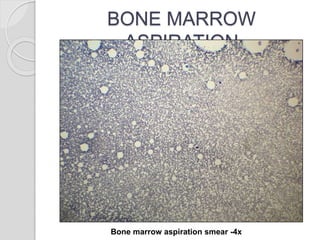
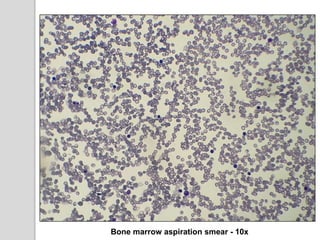
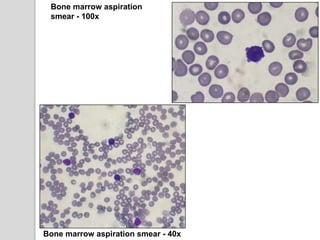
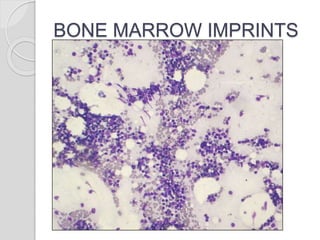
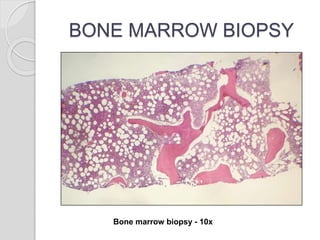
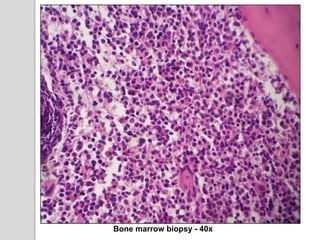
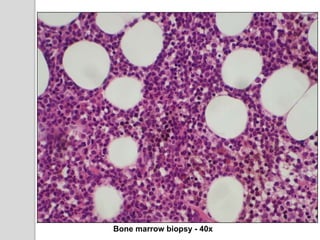
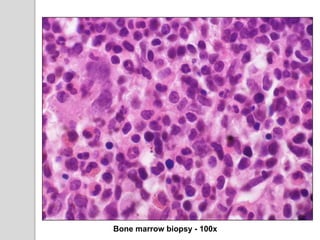
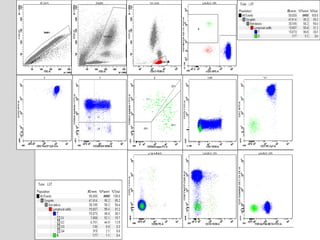
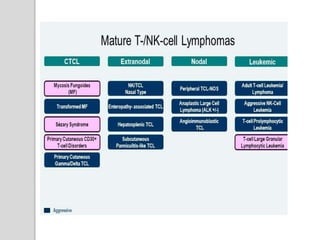
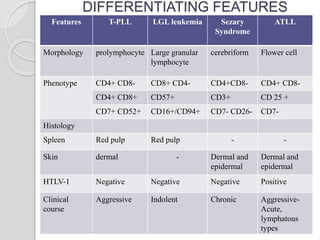
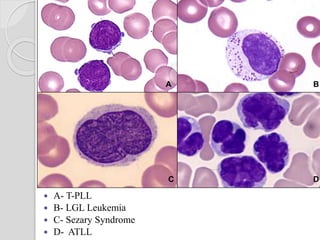
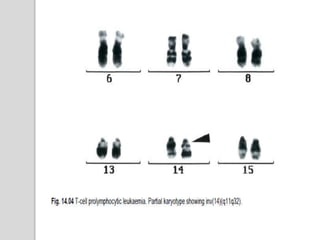
This document presents a case study of a 42-year-old male who presented with abdominal pain and was found to have a markedly elevated lymphocyte count on blood tests. Further testing revealed the lymphocytes to be abnormal in morphology. Bone marrow biopsies and additional testing led to a diagnosis of T-cell prolymphocytic leukemia (T-PLL), a rare and aggressive form of T-cell lymphoma. The document discusses the clinical features, distinguishing characteristics from other lymphomas, molecular genetics, disease progression, and poor prognosis of T-PLL.






















































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)





