Download to read offline





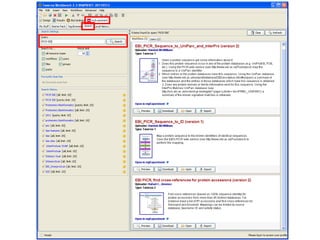


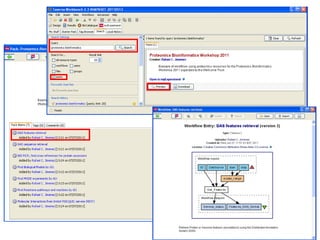


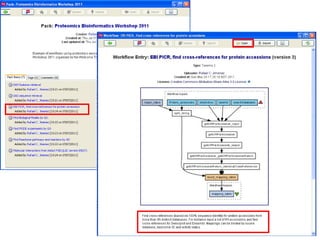
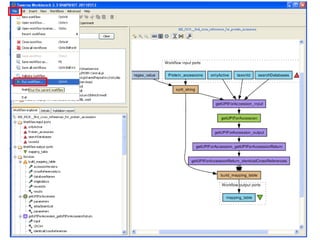
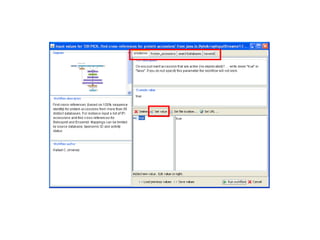
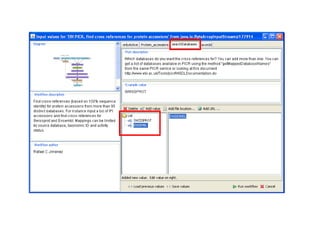
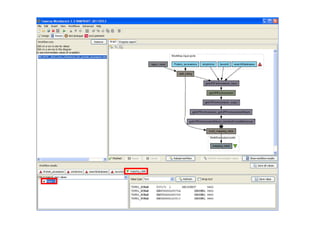





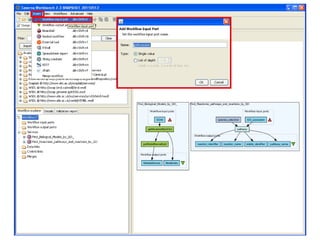
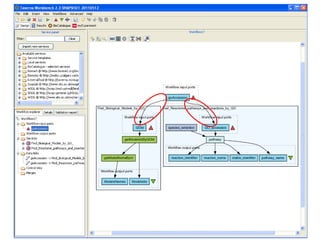
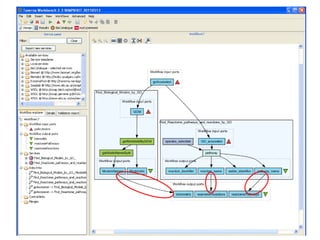
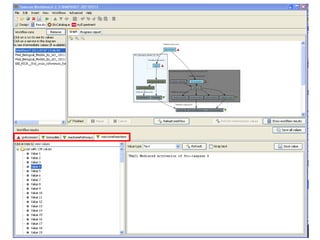



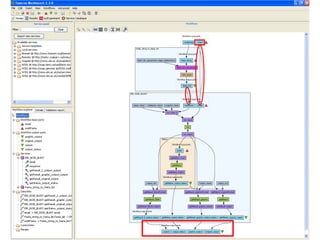





This document provides instructions for a series of exercises using the Taverna workflow management system. The exercises include searching for workflows on MyExperiment, running workflows to map protein accessions and perform BLAST searches, building a nested workflow, and running workflows from the command line. The goal is to familiarize users with searching, running, and constructing workflows in Taverna.
































































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)


