
Recommended
More Related Content
What's hot
What's hot (20)
Similar to Sequence assembly
Similar to Sequence assembly (20)
More from Dr. Naveen Gaurav srivastava
More from Dr. Naveen Gaurav srivastava (20)
Recently uploaded
Recently uploaded (20)
Sequence assembly
- 1. Genome Sequence Assembly Dr. Naveen Gaurav Associate Professor and Head Department of Biotechnology Shri Guru Ram Rai University Dehradun
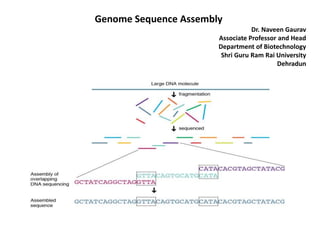
- 2. Genome Sequence Assembly Sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. This is needed as DNA sequencing technology cannot read whole genomes in one go, but rather reads small pieces of between 20 and 30,000 bases, depending on the technology used. Typically the short fragments, called reads, result from shotgun sequencing genomic DNA, or gene transcript (ESTs). The problem of sequence assembly can be compared to taking many copies of a book, passing each of them through a shredder with a different cutter, and piecing the text of the book back together just by looking at the shredded pieces. Besides the obvious difficulty of this task, there are some extra practical issues: the original may have many repeated paragraphs, and some shreds may be modified during shredding to have typos. Excerpts from another book may also be added in, and some shreds may be completely unrecognizable. Genome assemblers The first sequence assemblers began to appear in the late 1980s and early 1990s as variants of simpler sequence alignment programs to piece together vast quantities of fragments generated by automated sequencing instruments called DNA sequencers. As the sequenced organisms grew in size and complexity (from small viruses over plasmids to bacteria and finally eukaryotes), the assembly programs used in these genome projects needed increasingly sophisticated strategies to handle: 1. terabytes of sequencing data which need processing on computing clusters; 2. identical and nearly identical sequences (known as repeats) which can, in the worst case, 3. increase the time and space complexity of algorithms quadratically;
- 3. DNA read errors in the fragments from the sequencing instruments, which can confound assembly. Faced with the challenge of assembling the first larger eukaryotic genomes—the fruit fly Drosophila melanogaster in 2000 and the human genome just a year later,—scientists developed assemblers like Celera Assembler and Arachne able to handle genomes of 130 million (e.g., the fruit fly D. melanogaster) to 3 billion (e.g., the human genome) base pairs. Subsequent to these efforts, several other groups, mostly at the major genome sequencing centers, built large-scale assemblers, and an open source effort known as AMOS was launched to bring together all the innovations in genome assembly technology under the open source framework. Strategy how a sequence assembler would take fragments (shown below the black bar) and match overlaps among them to assembly the final sequence (in black). Potentially problematic repeats are shown above the sequence (in pink above). Without overlapping fragments it may be impossible to assign these segments to any specific region.
- 4. EST assemblers Expressed sequence tag or EST assembly was an early strategy, dating from the mid-1990s to the mid-2000s, to assemble individual genes rather than whole genomes. The problem differs from genome assembly in several ways. The input sequences for EST assembly are fragments of the transcribed mRNA of a cell and represent only a subset of the whole genome. A number of algorithmical problems differ between genome and EST assembly. For instance, genomes often have large amounts of repetitive sequences, concentrated in the intergenic regions. Transcribed genes contain many fewer repeats, making assembly somewhat easier. On the other hand, some genes are expressed (transcribed) in very high numbers (e.g., housekeeping genes), which means that unlike whole-genome shotgun sequencing, the reads are not uniformly sampled across the genome. EST assembly is made much more complicated by features like (cis-) alternative splicing, trans-splicing, single-nucleotide polymorphism, and post-transcriptional modification. Beginning in 2008 when RNA-Seq was invented, EST sequencing was replaced by this far more efficient technology, described under de novo transcriptome assembly. De-novo vs. mapping assembly In sequence assembly, two different types can be distinguished: 1. de-novo: assembling short reads to create full-length (sometimes novel) sequences, without using a template (see de novo sequence assemblers, de novo transcriptome assembly) 2. mapping: assembling reads against an existing backbone sequence, building a sequence that is similar but not necessarily identical to the backbone sequence
- 5. In terms of complexity and time requirements, de-novo assemblies are orders of magnitude slower and more memory intensive than mapping assemblies. This is mostly due to the fact that the assembly algorithm needs to compare every read with every other read (an operation that has a naive time complexity of O(n2). Referring to the comparison drawn to shredded books in the introduction: while for mapping assemblies one would have a very similar book as a template (perhaps with the names of the main characters and a few locations changed), de-novo assemblies present a more daunting challenge in that one would not know beforehand whether this would become a science book, a novel, a catalogue, or even several books. Also, every shred would be compared with every other shred. Handling repeats in de-novo assembly requires the construction of a graph representing neighboring repeats. Such information can be derived from reading a long fragment covering the repeats in full or only its two ends. On the other hand, in a mapping assembly, parts with multiple or no matches are usually left for another assembling technique to look into. Influence of technological changes The complexity of sequence assembly is driven by two major factors: the number of fragments and their lengths. While more and longer fragments allow better identification of sequence overlaps, they also pose problems as the underlying algorithms show quadratic or even exponential complexity behaviour to both number of fragments and their length. And while shorter sequences are faster to align, they also complicate the layout phase of an assembly as shorter reads are more difficult to use with repeats or near identical repeats.
- 6. n the earliest days of DNA sequencing, scientists could only gain a few sequences of short length (some dozen bases) after weeks of work in laboratories. Hence, these sequences could be aligned in a few minutes by hand. In 1975, the dideoxy termination method (AKA Sanger sequencing) was invented and until shortly after 2000, the technology was improved up to a point where fully automated machines could churn out sequences in a highly parallelised mode 24 hours a day. Large genome centers around the world housed complete farms of these sequencing machines, which in turn led to the necessity of assemblers to be optimised for sequences from whole-genome shotgun sequencing projects where the reads 1. are about 800–900 bases long 2. contain sequencing artifacts like sequencing and cloning vectors 3. have error rates between 0.5 and 10% With the Sanger technology, bacterial projects with 20,000 to 200,000 reads could easily be assembled on one computer. Larger projects, like the human genome with approximately 35 million reads, needed large computing farms and distributed computing. By 2004 / 2005, pyrosequencing had been brought to commercial viability by 454 Life Sciences. This new sequencing method generated reads much shorter than those of Sanger sequencing: initially about 100 bases, now 400-500 bases. Its much higher throughput and lower cost (compared to Sanger sequencing) pushed the adoption of this technology by genome centers, which in turn pushed development of sequence assemblers that could efficiently handle the read sets. The sheer amount of data coupled with technology-specific error patterns in the reads delayed development of assemblers; at the beginning in 2004 only the Newbler assembler from 454 was available. Released in mid-2007, the hybrid version of the MIRA assembler by Chevreux et al. was the first freely available assembler that could assemble 454 reads as well as mixtures of 454 reads and Sanger reads. Assembling sequences from different sequencing technologies was subsequently coined hybrid assembly.
- 7. From 2006, the Illumina (previously Solexa) technology has been available and can generate about 100 million reads per run on a single sequencing machine. Compare this to the 35 million reads of the human genome project which needed several years to be produced on hundreds of sequencing machines. Illumina was initially limited to a length of only 36 bases, making it less suitable for de novo assembly (such as de novo transcriptome assembly), but newer iterations of the technology achieve read lengths above 100 bases from both ends of a 3-400bp clone. Announced at the end of 2007, the SHARCGS assemblerby Dohm et al. was the first published assembler that was used for an assembly with Solexa reads. It was quickly followed by a number of others. Later, new technologies like SOLiD from Applied Biosystems, Ion Torrent and SMRT were released and new technologies (e.g. Nanopore sequencing) continue to emerge. Despite the higher error rates of these technologies they are important for assembly because their longer read length helps to address the repeat problem. It is impossible to assemble through a perfect repeat that is longer than the maximum read length; however, as reads become longer the chance of a perfect repeat that large becomes small. This gives longer sequencing reads an advantage in assembling repeats even if they have low accuracy (~85%). Greedy algorithm Given a set of sequence fragments, the object is to find a longer sequence that contains all the fragments. Сalculate pairwise alignments of all fragments. Choose two fragments with the largest overlap. Merge chosen fragments. Repeat step 2 and 3 until only one fragment is left. The result need not be an optimal solution to the problem.
- 8. Thank you References: Online notes, notes from research papers and Books by google search Engine