Download to read offline
![Using Maestro and Gaussian 09 in the Qualitative analysis of
Endiynes (enyne-allenes)
Abstract
By Dr. Robert D. Craig,Ph.D.
-8,10,11 trihydroxy- 9- Bicyclo(7:2:2)undec 2- yne,4-ene,6-yne
Students in my group have carried out DFT and various Analytical techniques to study an
enyne-allene OR Enediyne- C11H5O4. Mapping the synthesize of C11H5O4 was done with alpha-
butanone. The FT-NMR (1
H and 13
C) and FT-Raman were obtained . The spectra was adequate to
analyze and were compared to literature values. The Mulliken, Lowdin, and NBO analysis were also
carried out on the enediynes. Students became familiar with DFT analysis , and using the molecule,
completed with respect each instrument (UV-VIS, FT-NMR, and FT-IR) using the B-3-YLP/6-311++(2p,3d),
MP2, and RHF-STO-3G-basis sets. The calculated HOMO and LUMO values were compared with spectra
taken on the Cary Fluorescence spectrophotometer.
Introduction
Ref 1
Enediynes undergo a Bergman cyclization reaction to form the labile 1,4-didehy-
drobenzene (p-benzyne) biradical. (1-3) The energetics of this reaction and the related
Schreiner–Pascal reaction as well as that of the Myers–Saito and Schmittel reactions of enyne-
allenes are discussed on the basis of a variety of quantum chemical and available experimental
results. (4-6)The computational investigation of enediynes has been beneficial for both
experimentalists and theoreticians because it has led to new synthetic challenges and new
computational methodologies. The computer-assisted drug design of new antitumor antibiotics
based on the biological activity of natural enediynes in now very popular for the understanding
of catalyzed enediyne reactions
Figure one shows Bicyclo(7:2:2)deca 2,4,6-yne-allene-8,10,11 triol -THIS IS J5-NEED 11 PREFIX
Bicyclo[7:2:2] triol molecule 72- C11H5O4 and Molecule 73- C17H11O4](https://image.slidesharecdn.com/qualitativeanddftanalysisofendiynes-140729111945-phpapp01/75/Qualitative-and-dft-analysis-of-endiynes-1-2048.jpg)




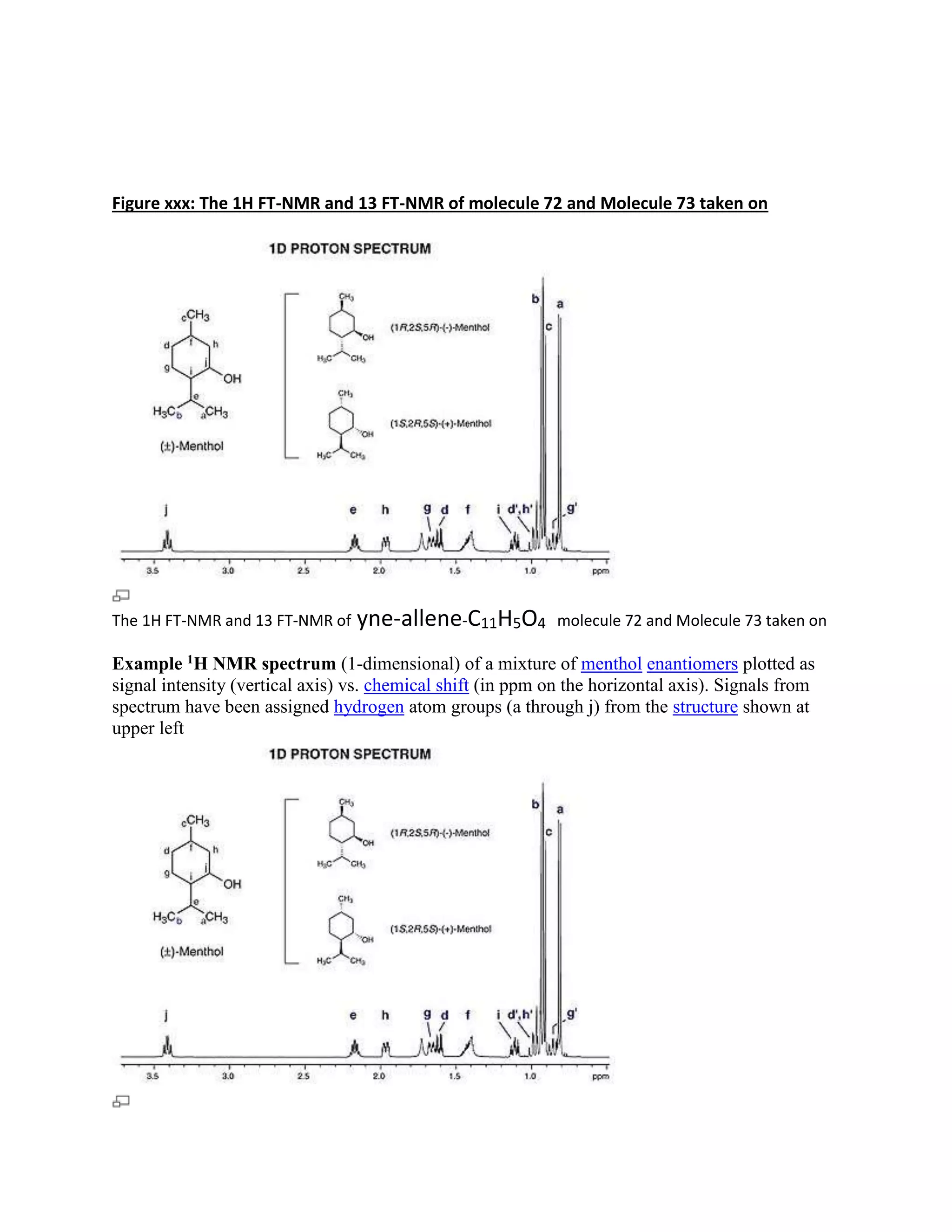



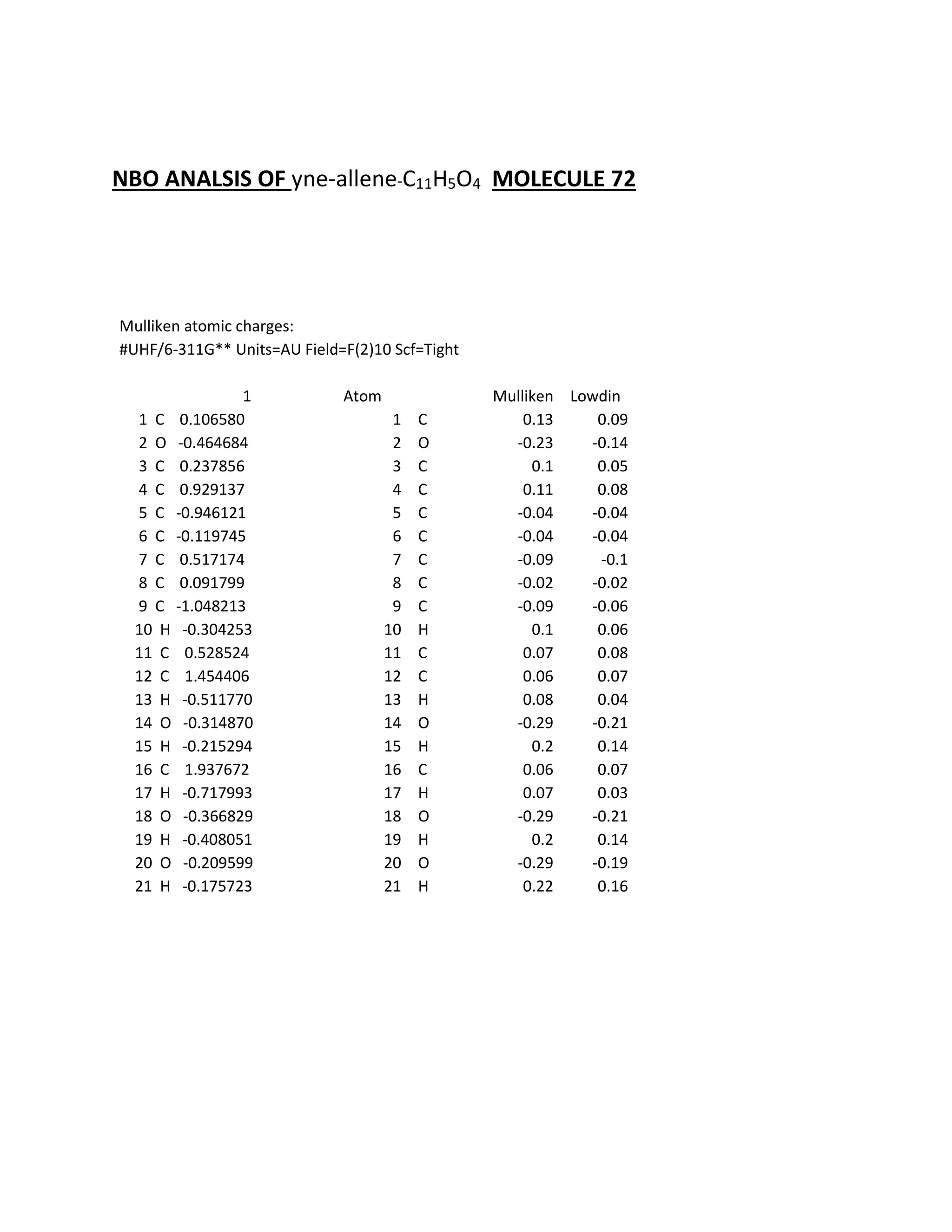




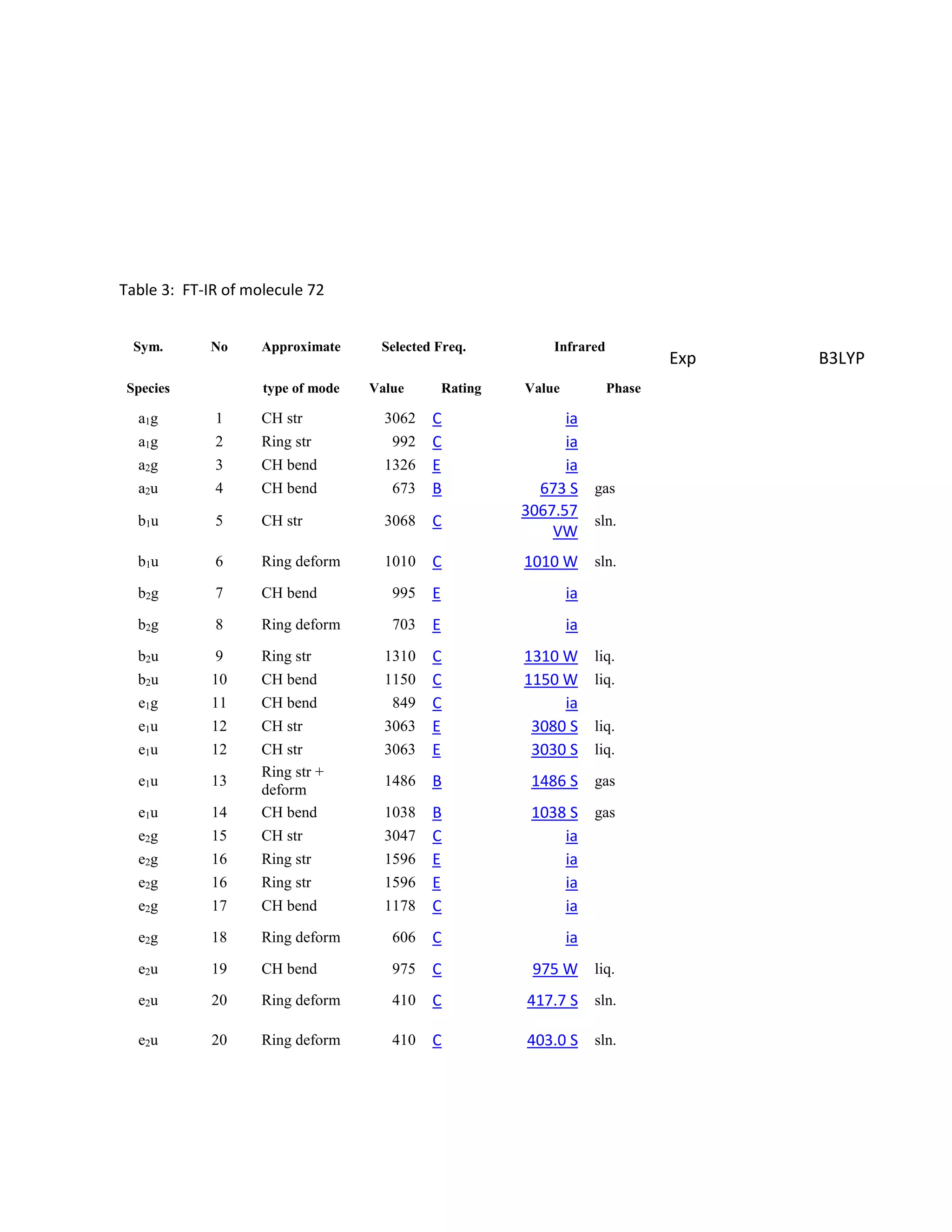




Students analyzed an enediyne compound, C11H5O4, using computational chemistry methods like DFT and various spectroscopic techniques. The FT-NMR (1H and 13C) and FT-Raman spectra were obtained and compared to literature values. DFT calculations using B3LYP, MP2, and RHF methods helped students understand the vibrational modes, NMR chemical shifts, and frontier molecular orbitals like HOMO and LUMO of the compound. Experimental UV-Vis and FT-IR spectra were also collected and analyzed. The computational results provided insight into the structural properties and reactivity of this biologically relevant enediyne compound.



































![Day%202%20 city%20college[1]](https://cdn.slidesharecdn.com/ss_thumbnails/day20220city20college1-140617185102-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)
![Sd1 wastewaterfactsheet[2]2](https://cdn.slidesharecdn.com/ss_thumbnails/sd1wastewaterfactsheet22-140617185132-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)















































