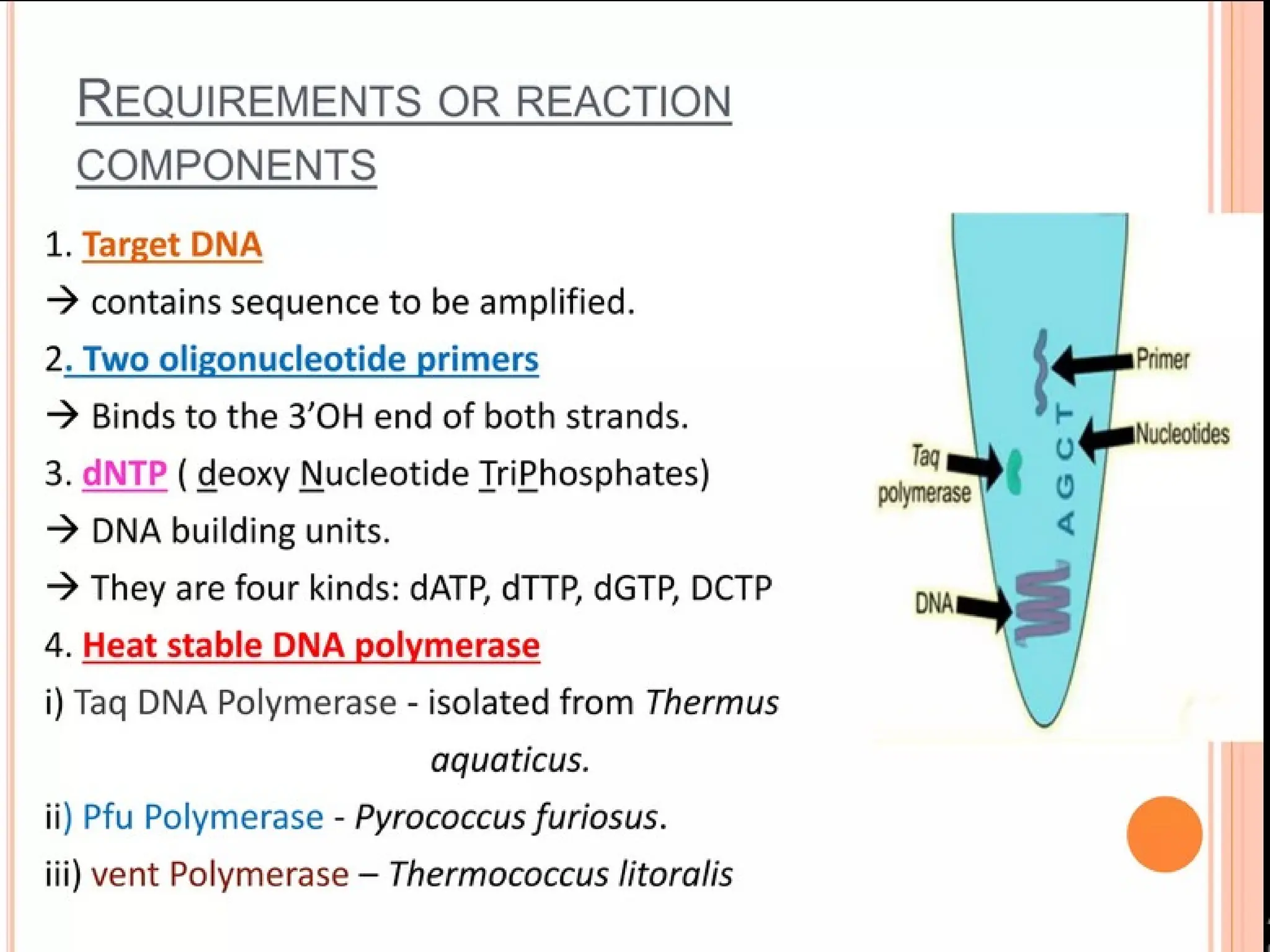
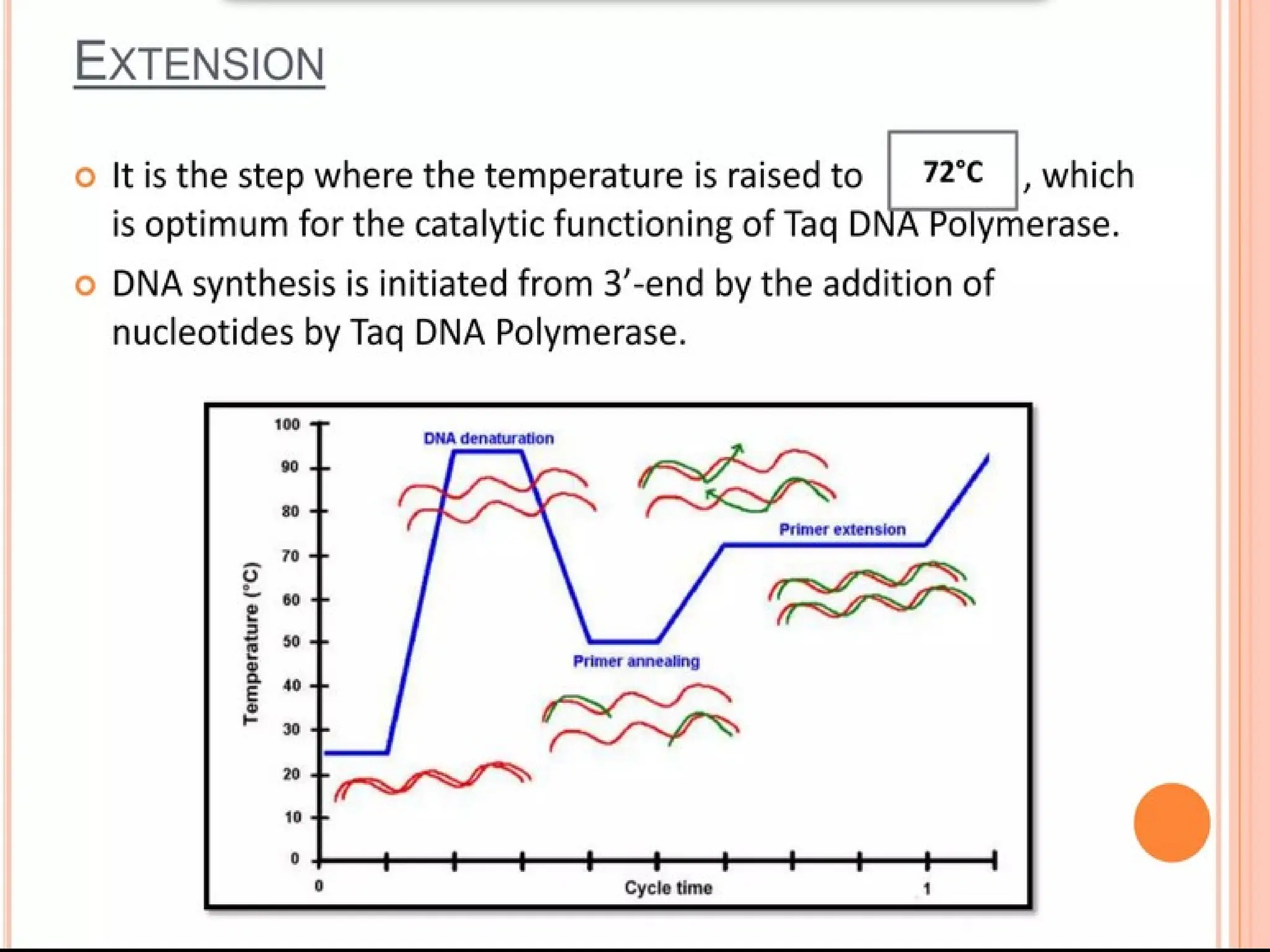
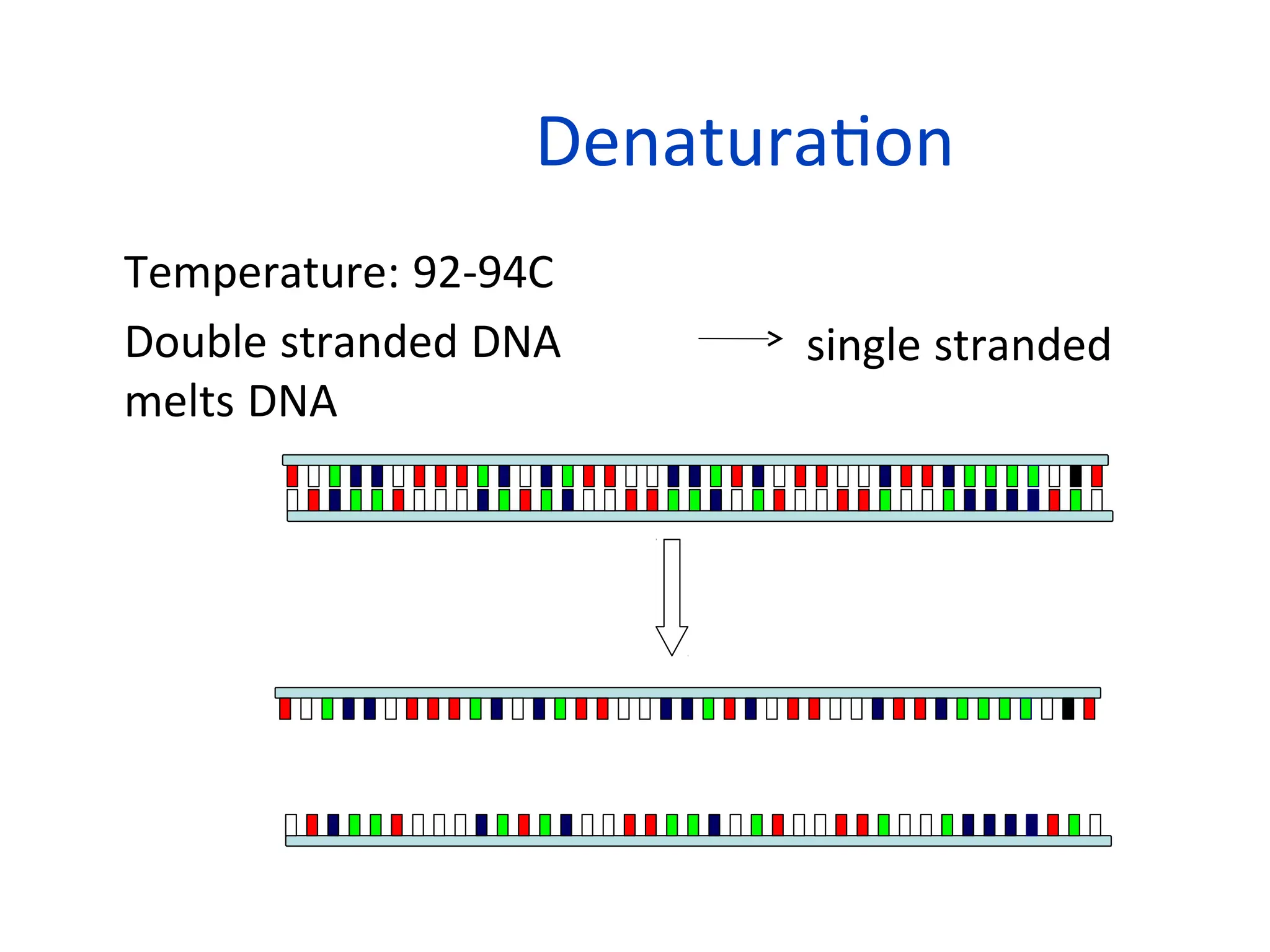
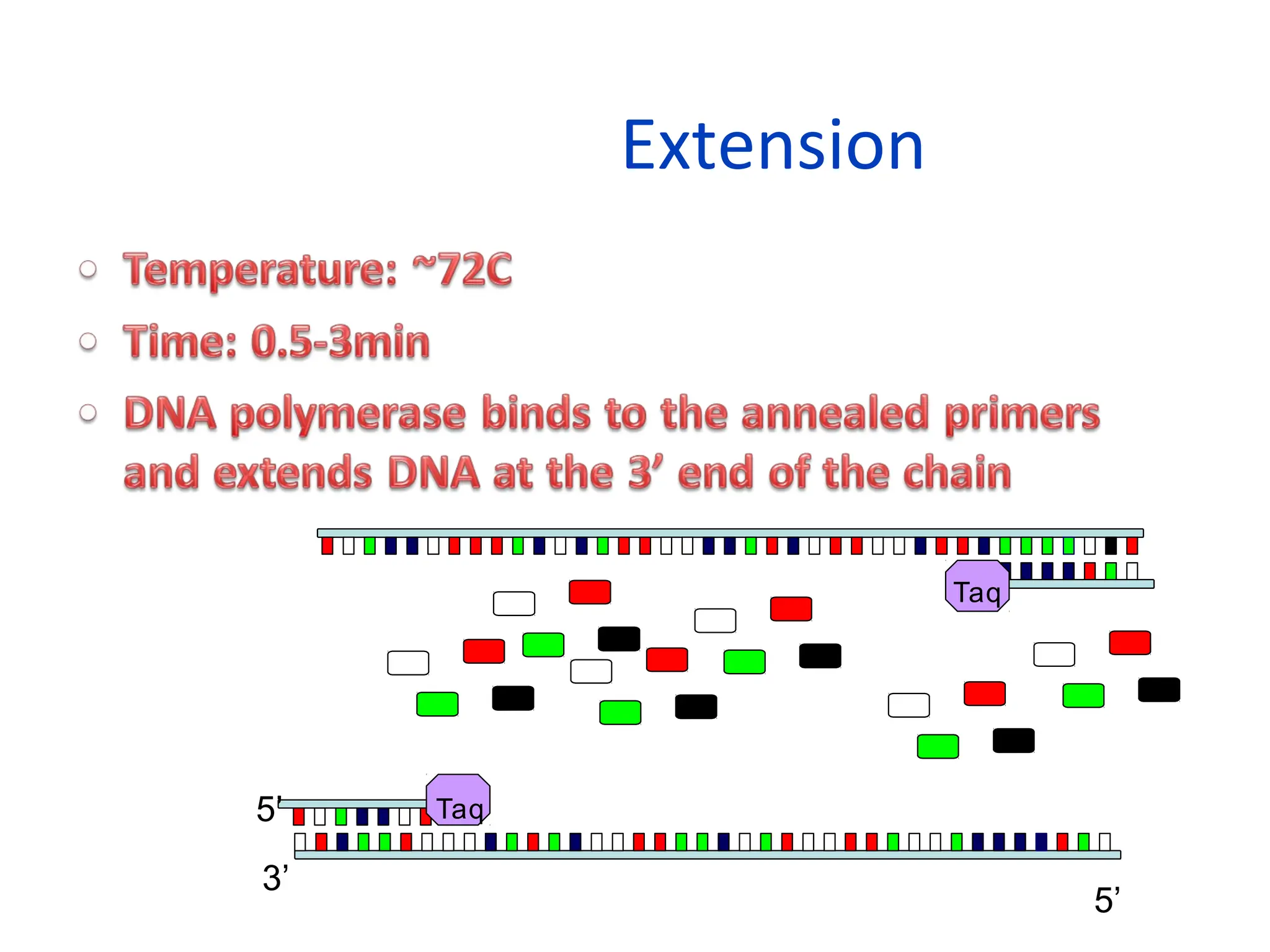
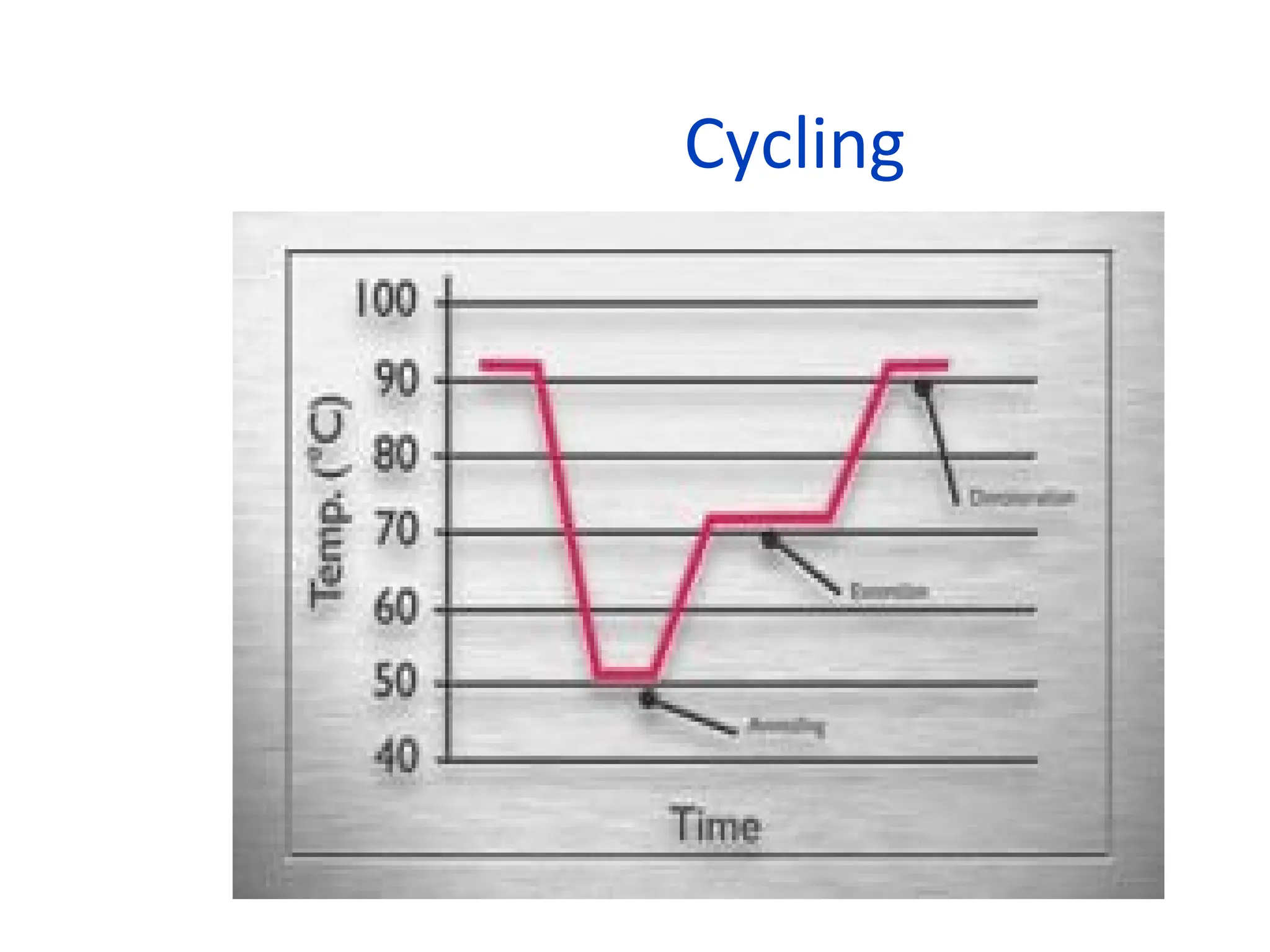
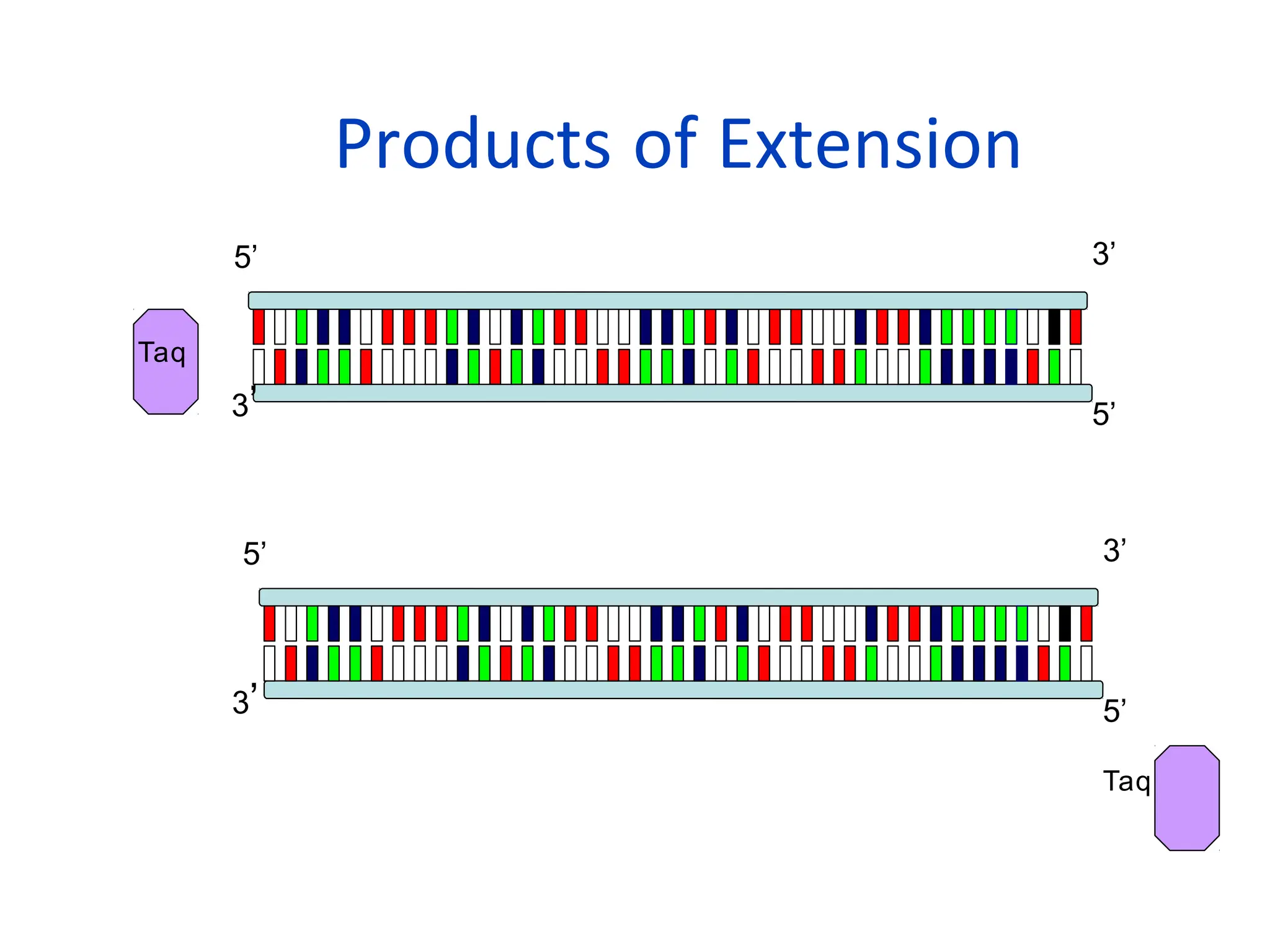
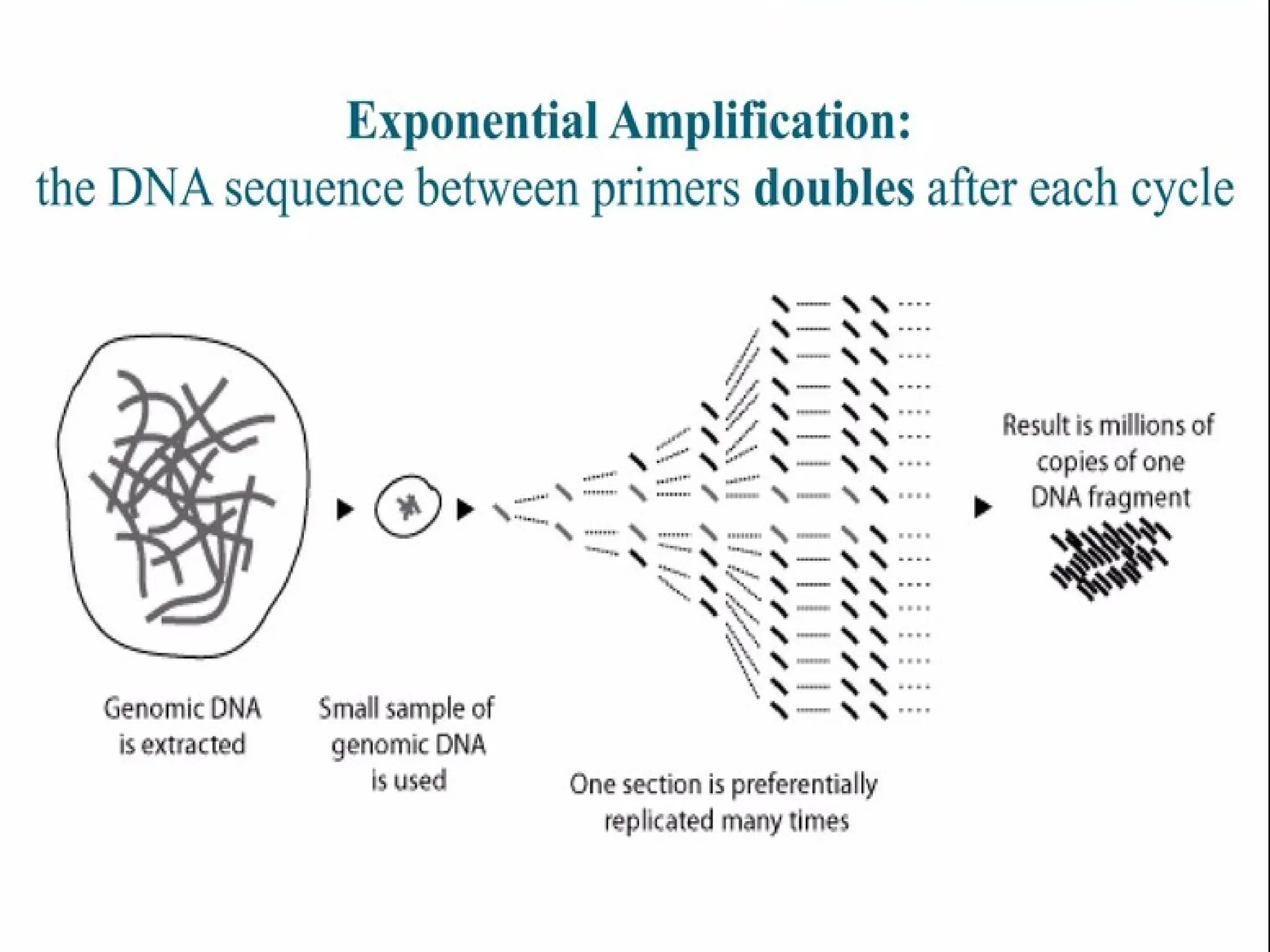
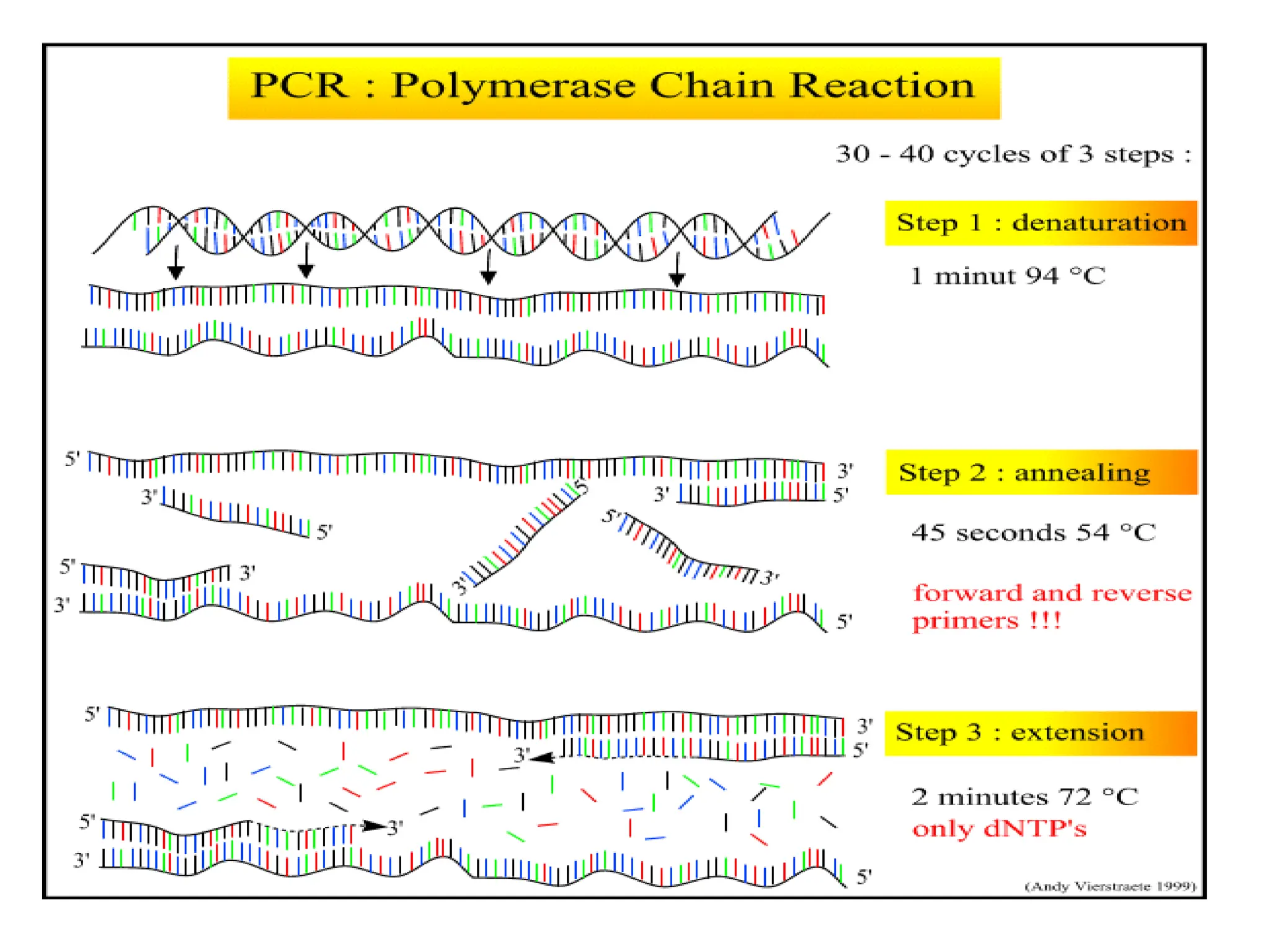
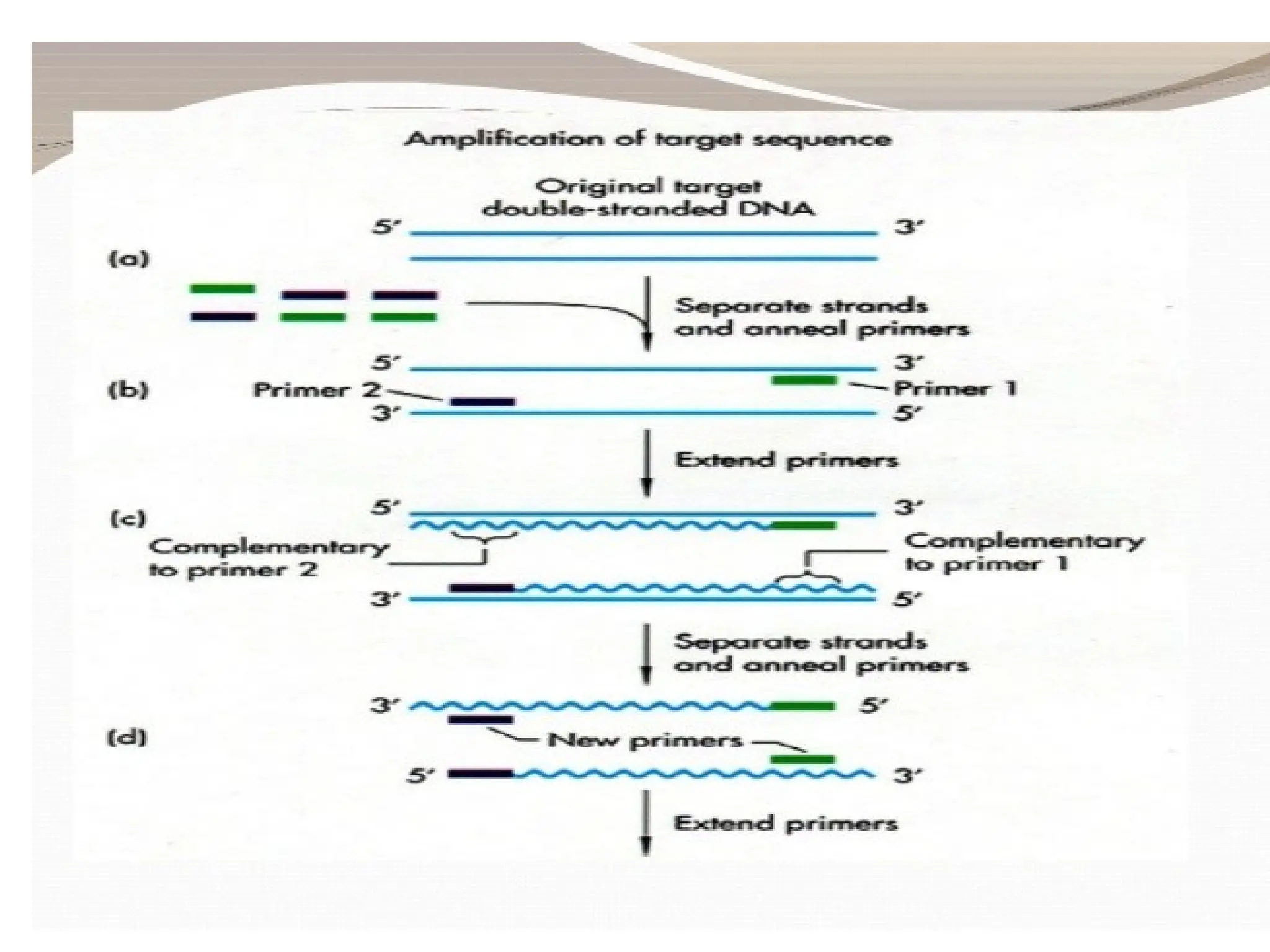
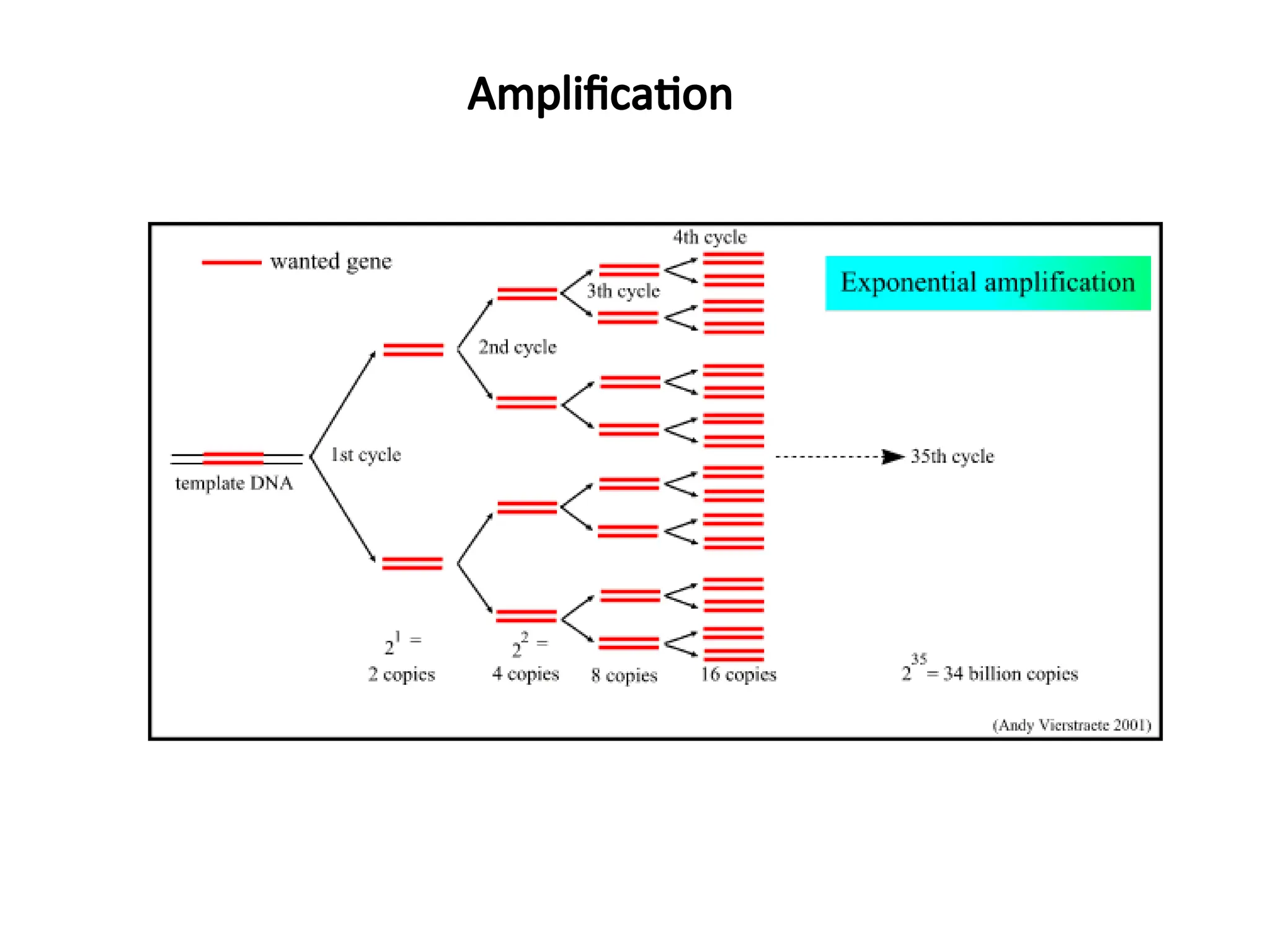
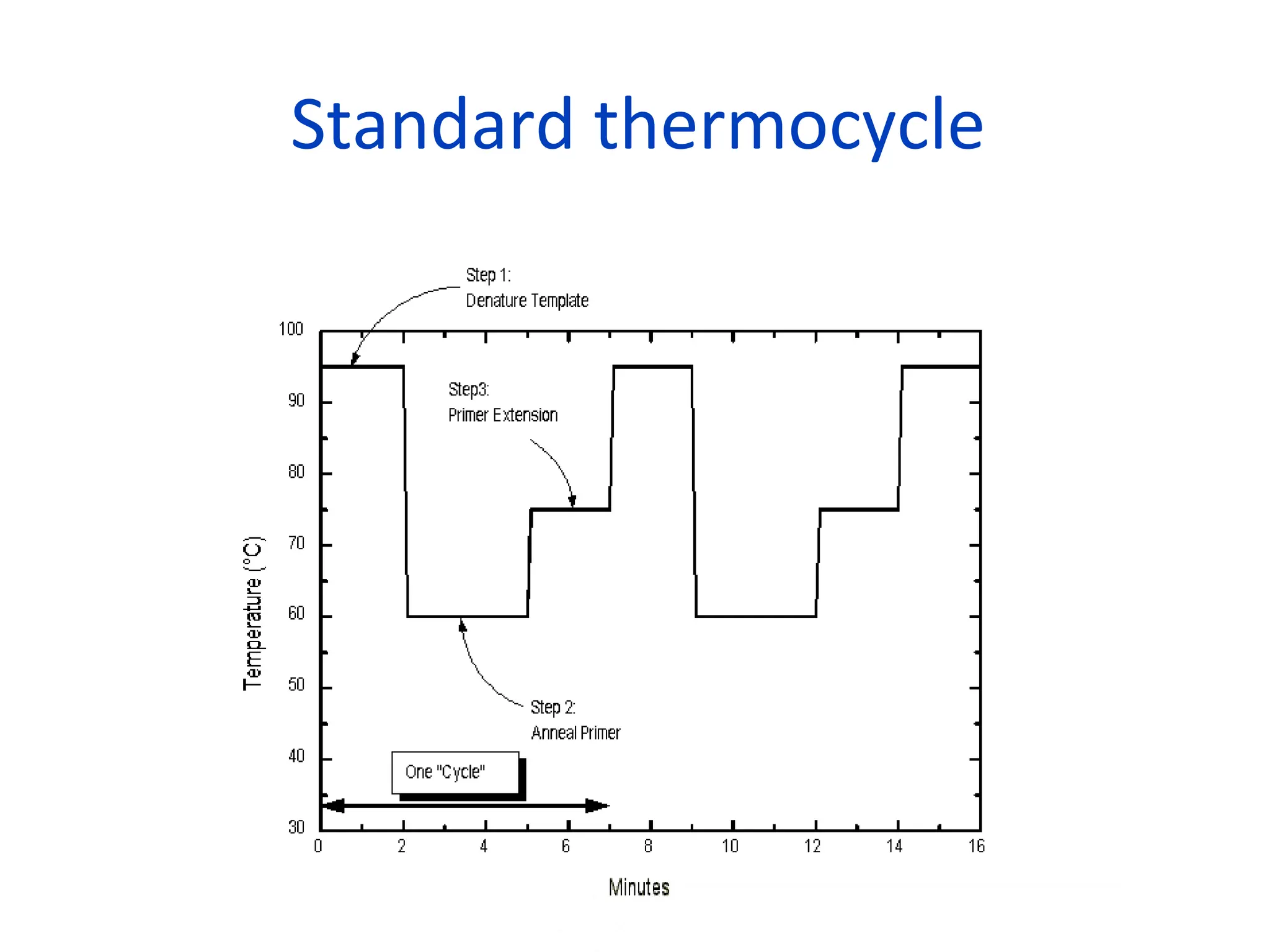
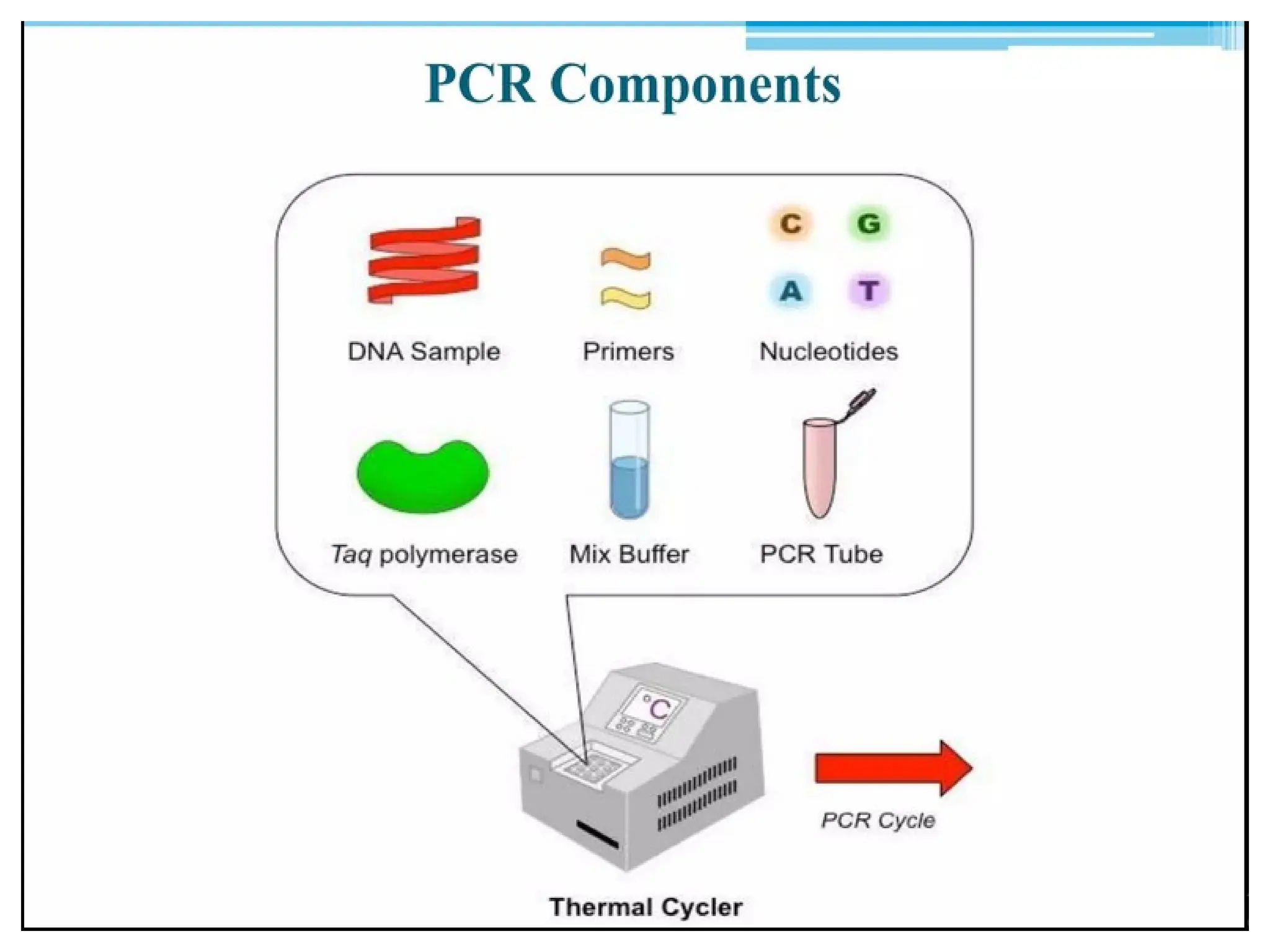
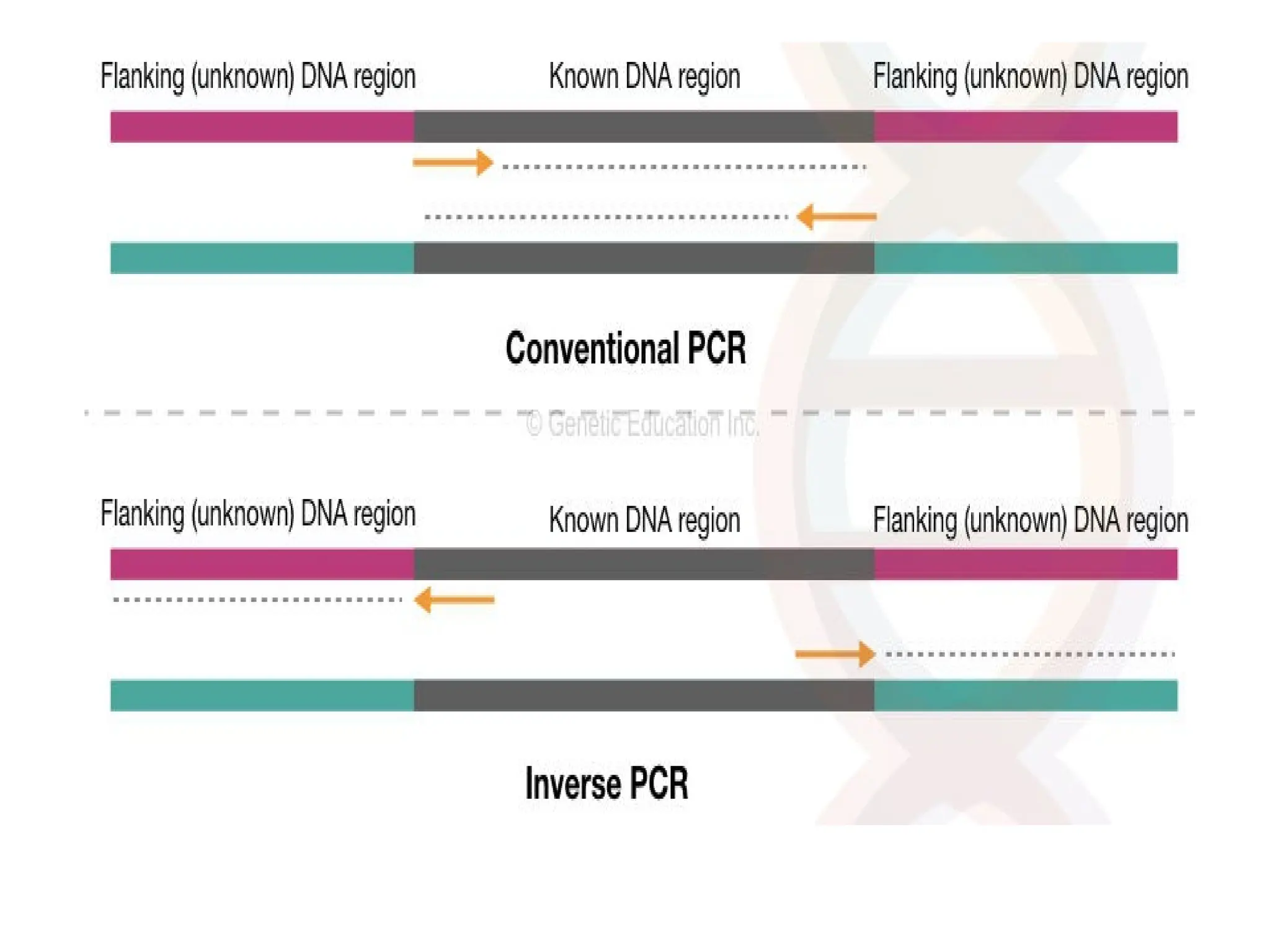
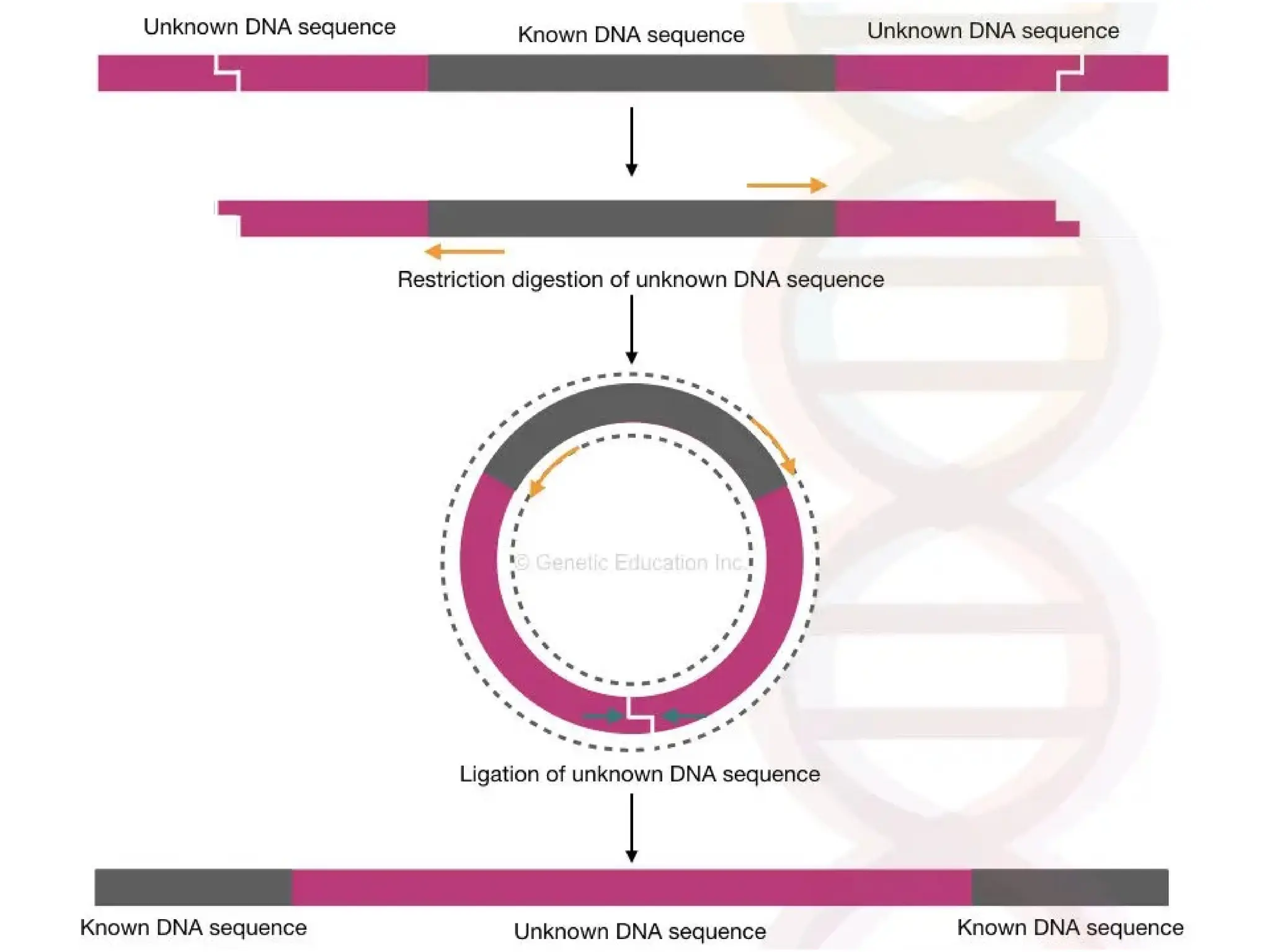
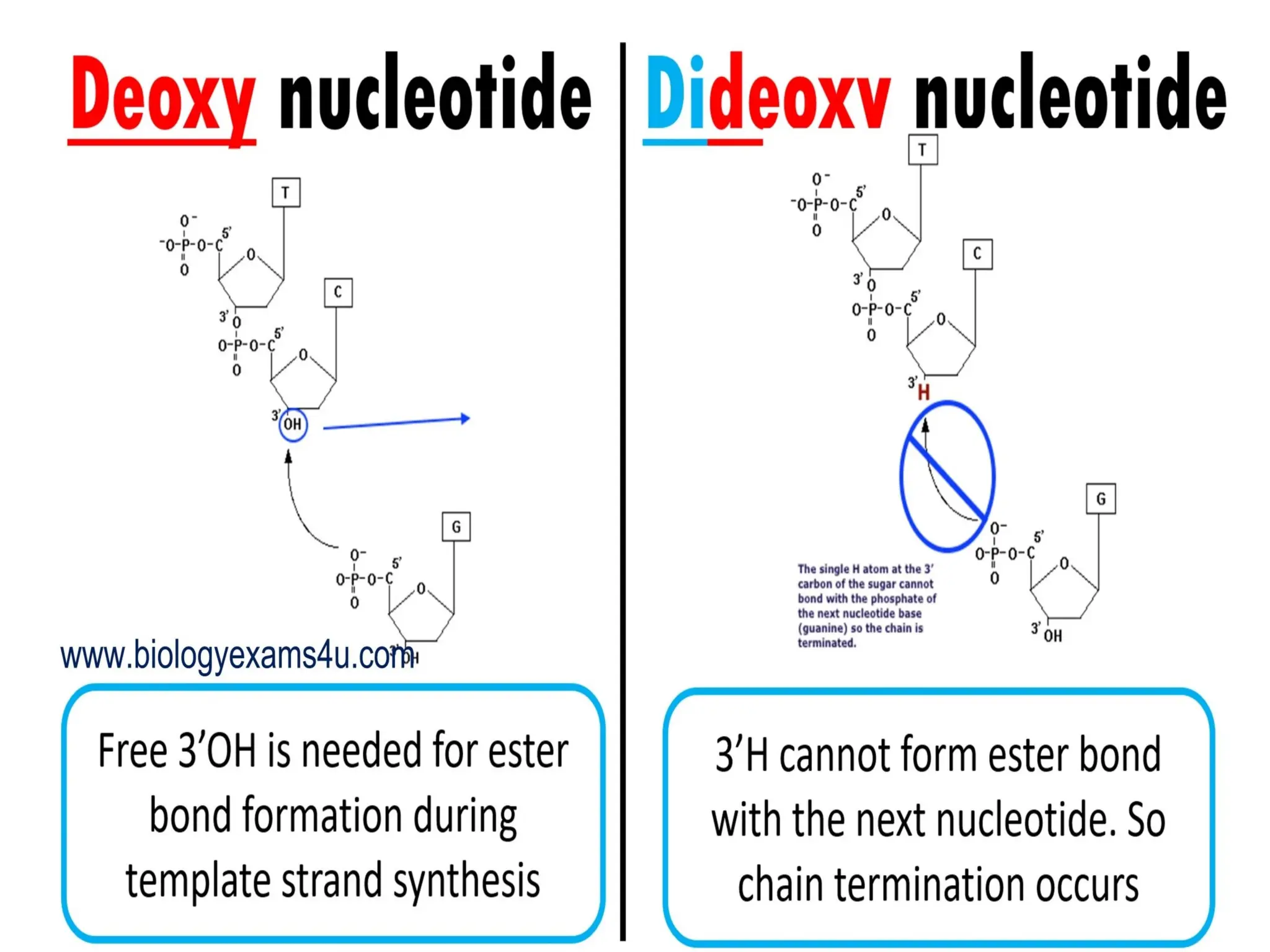
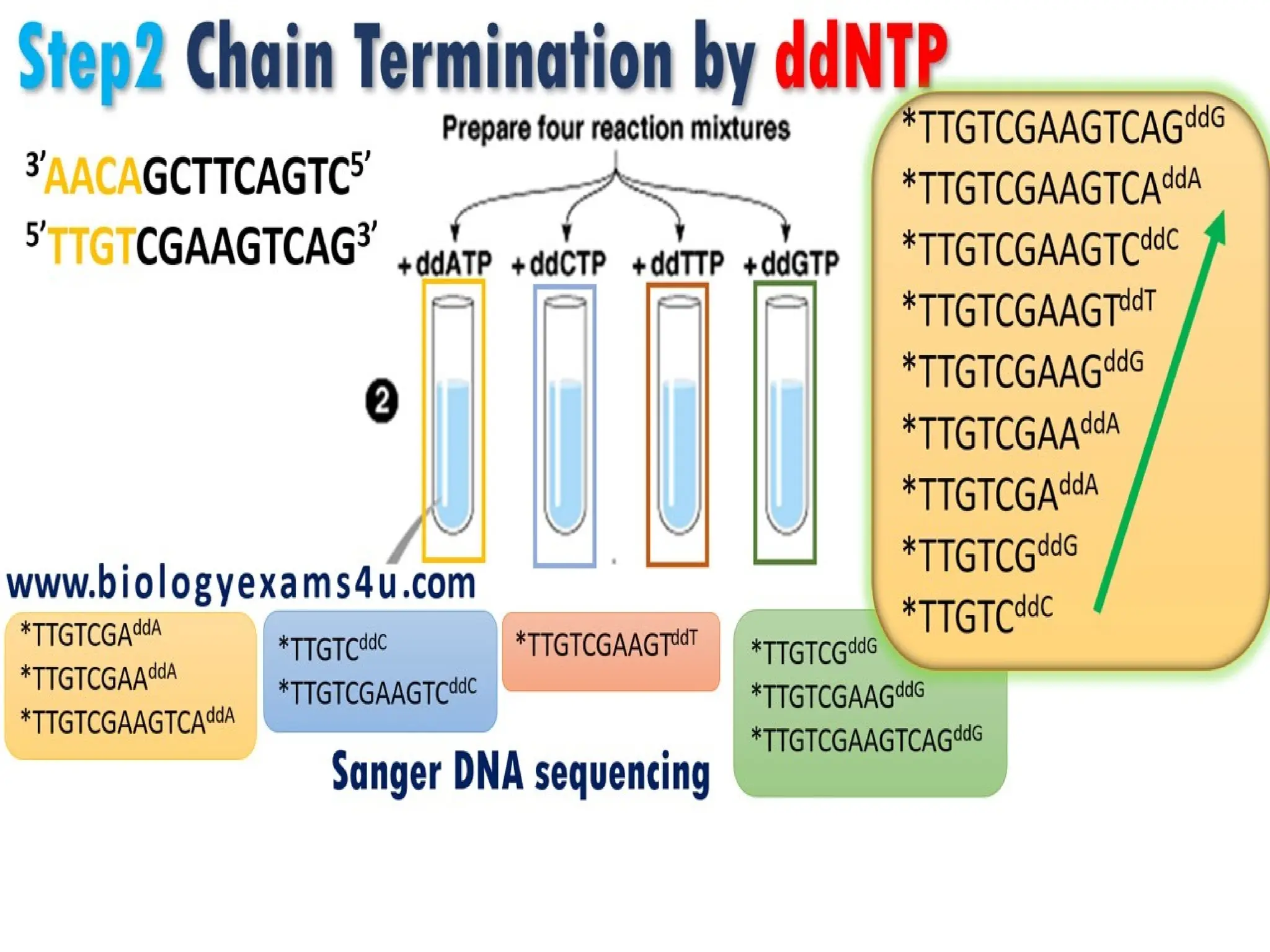
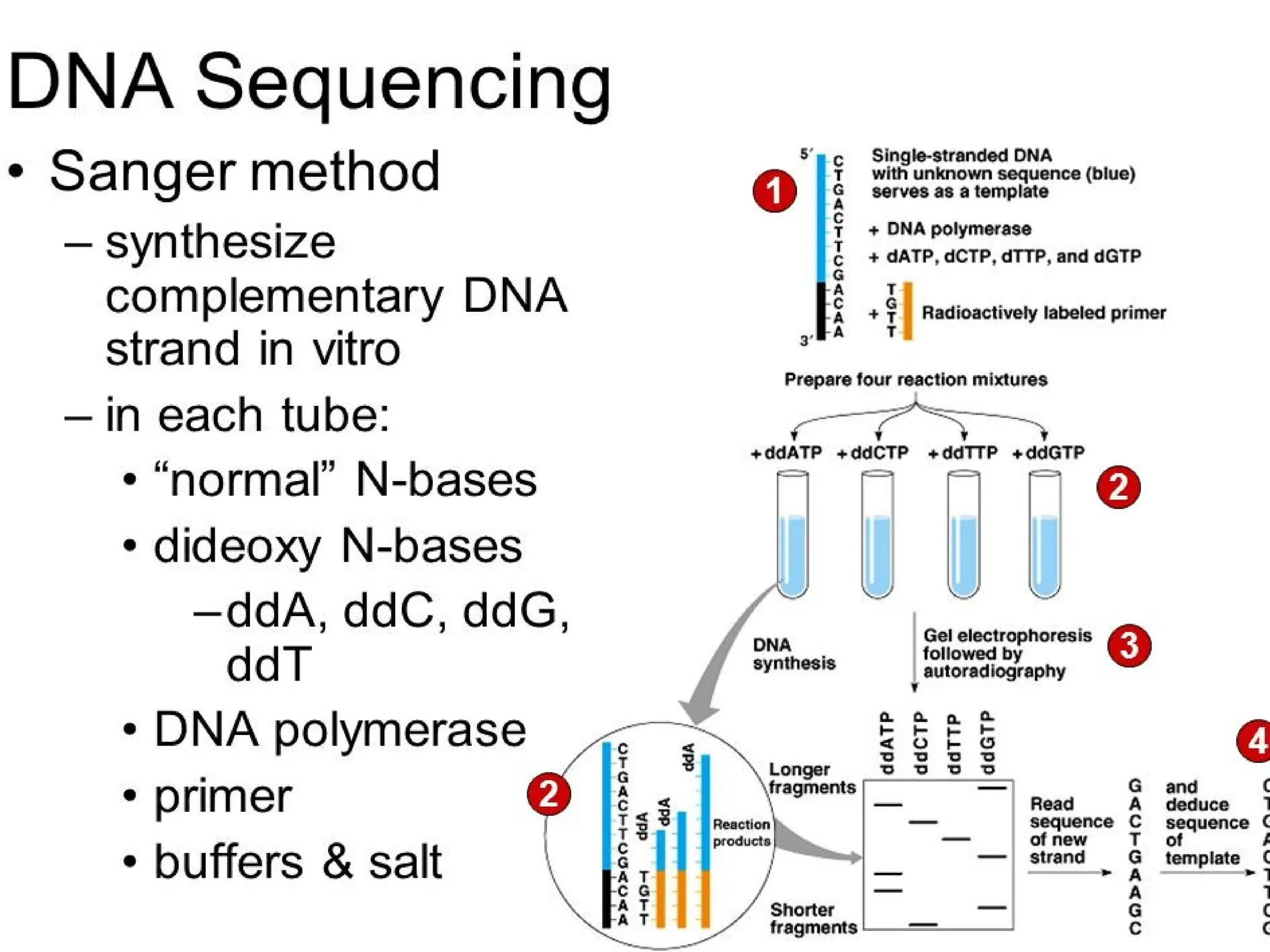
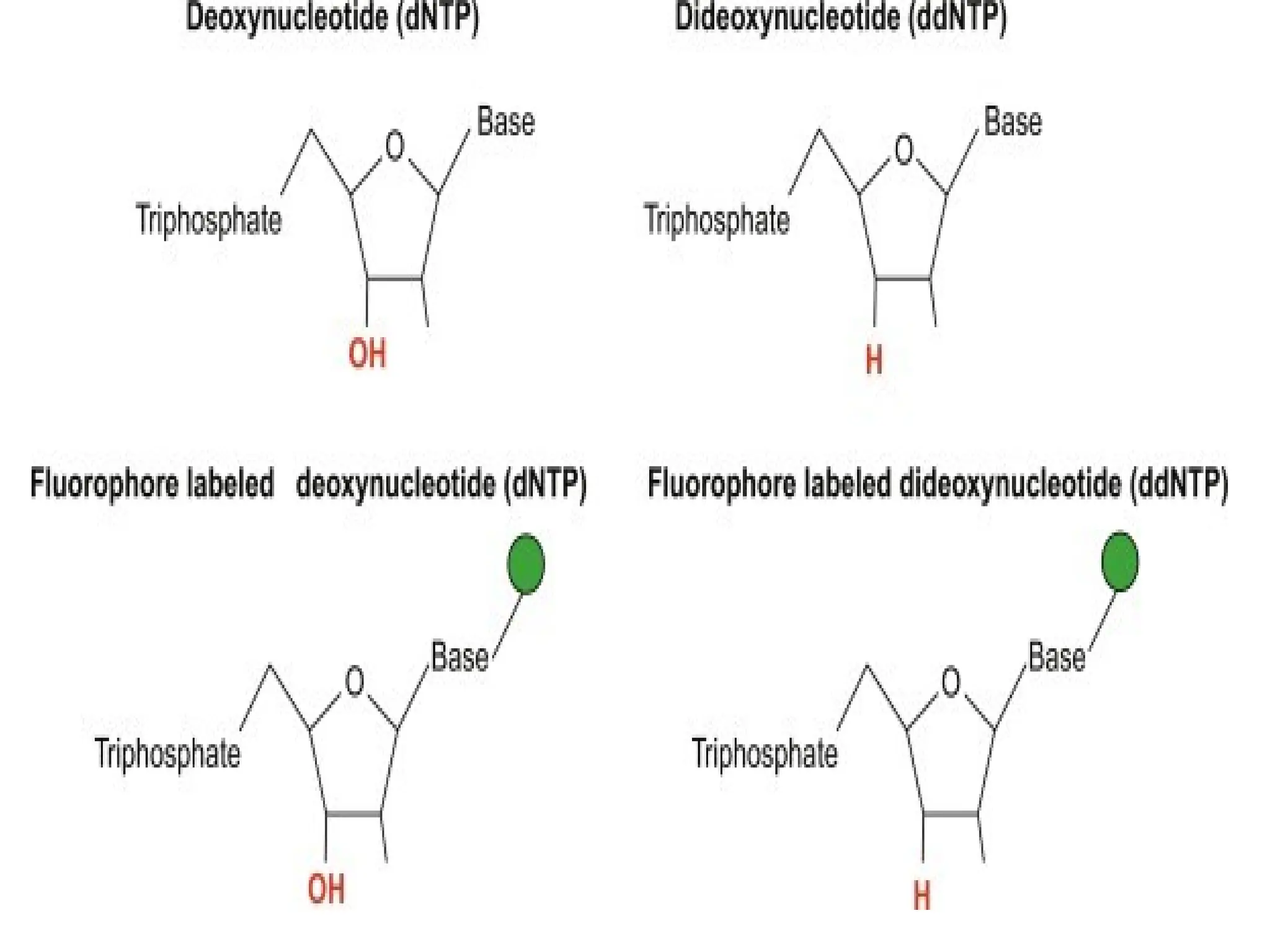
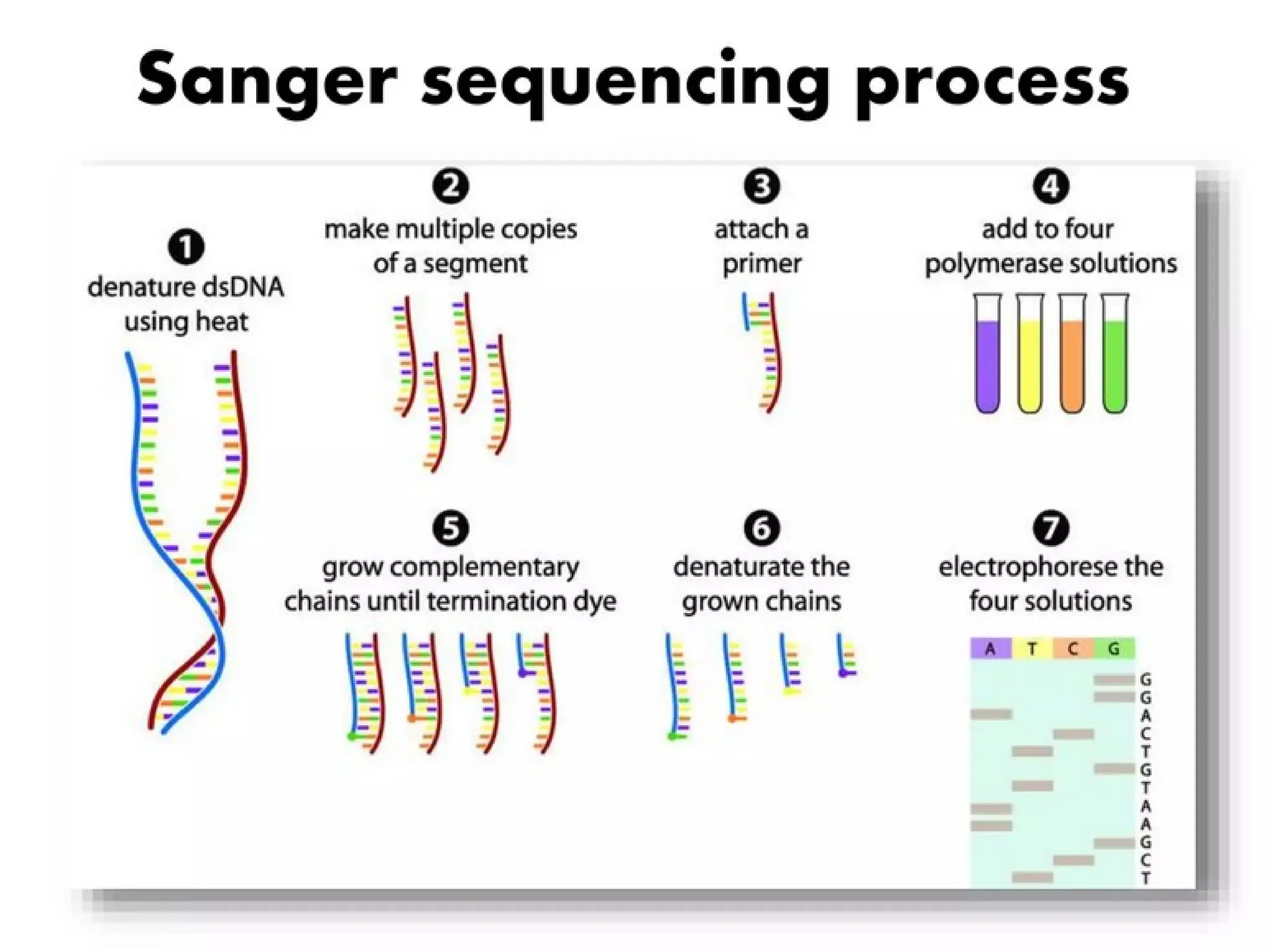
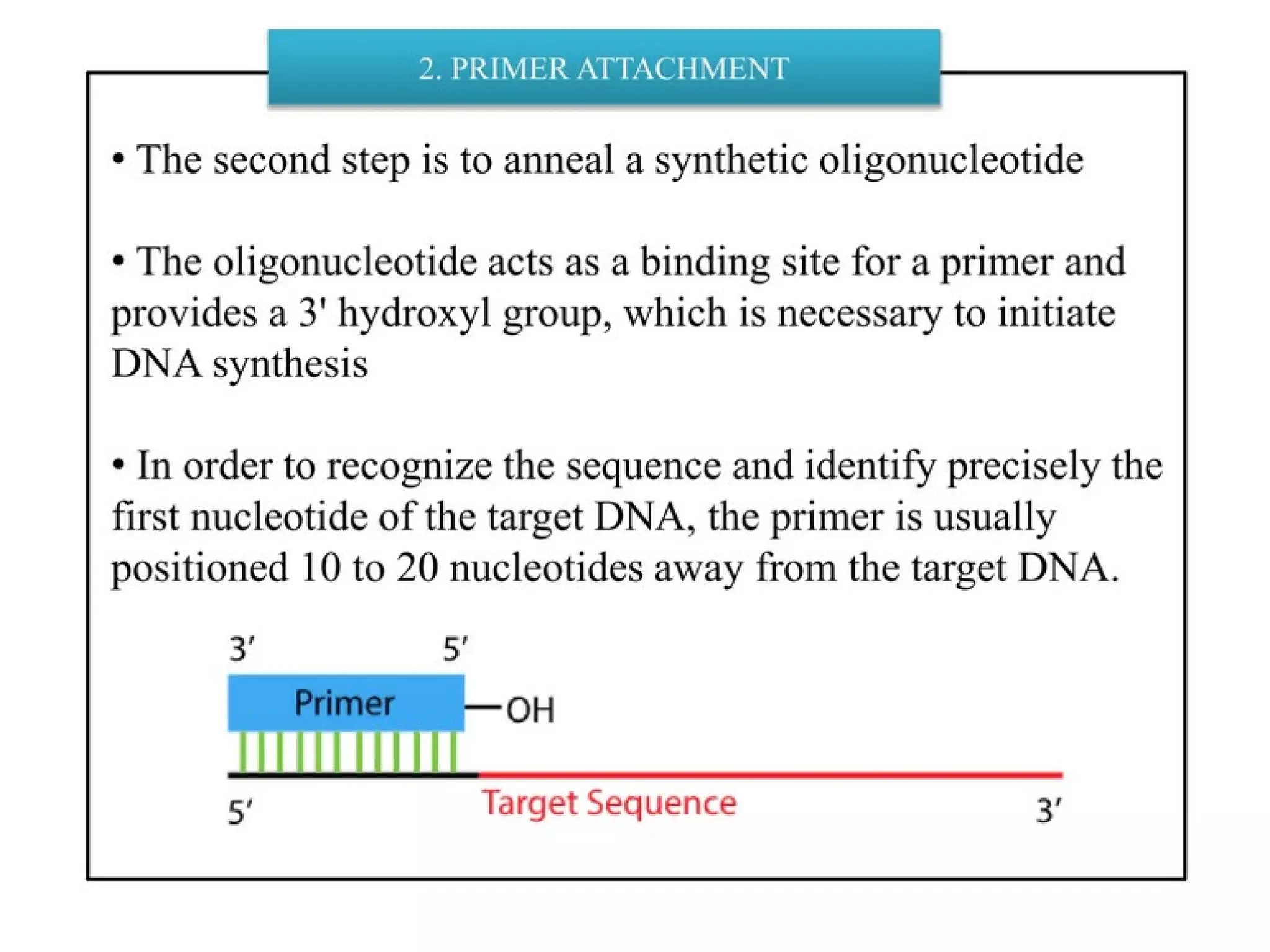
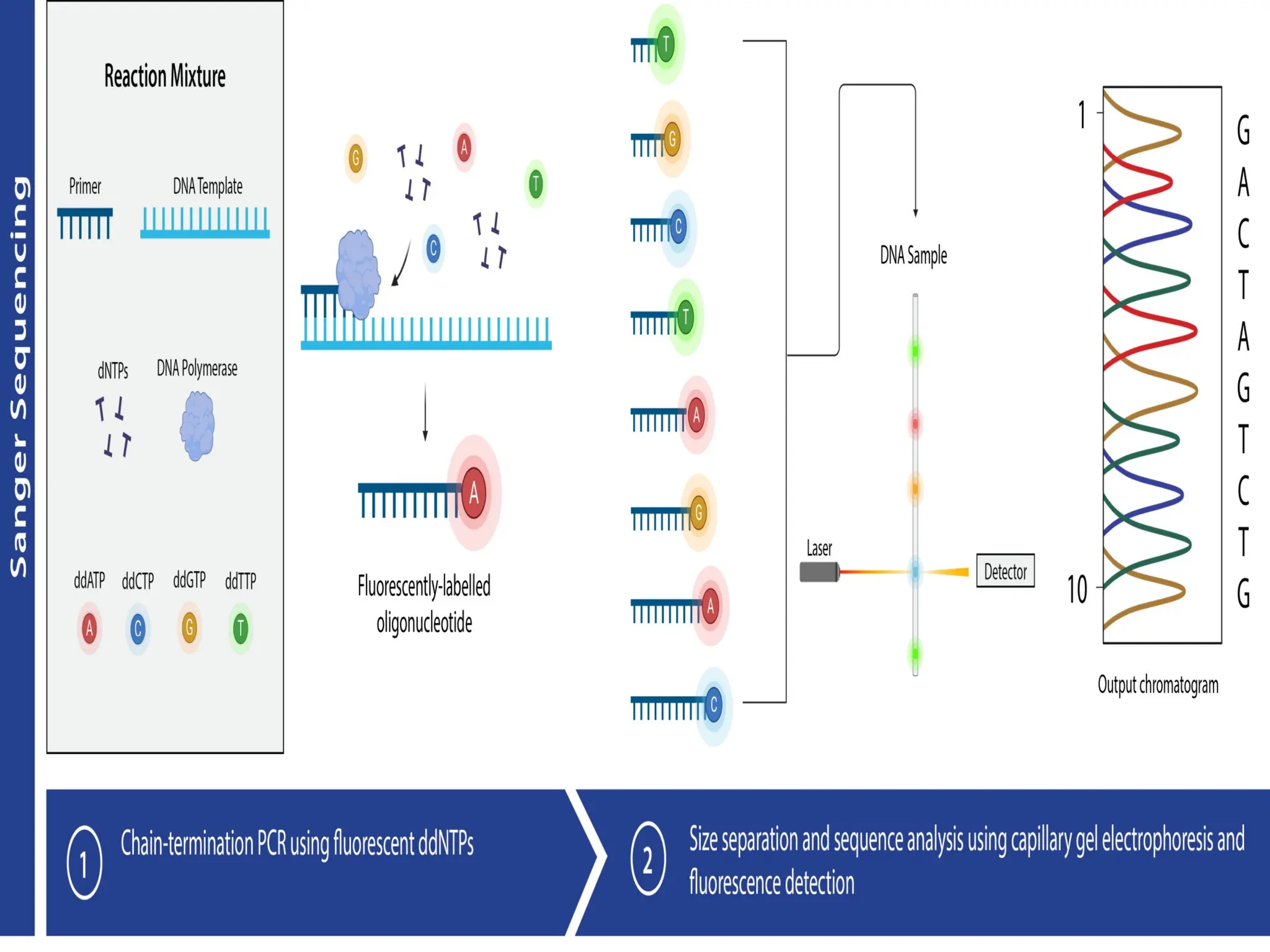
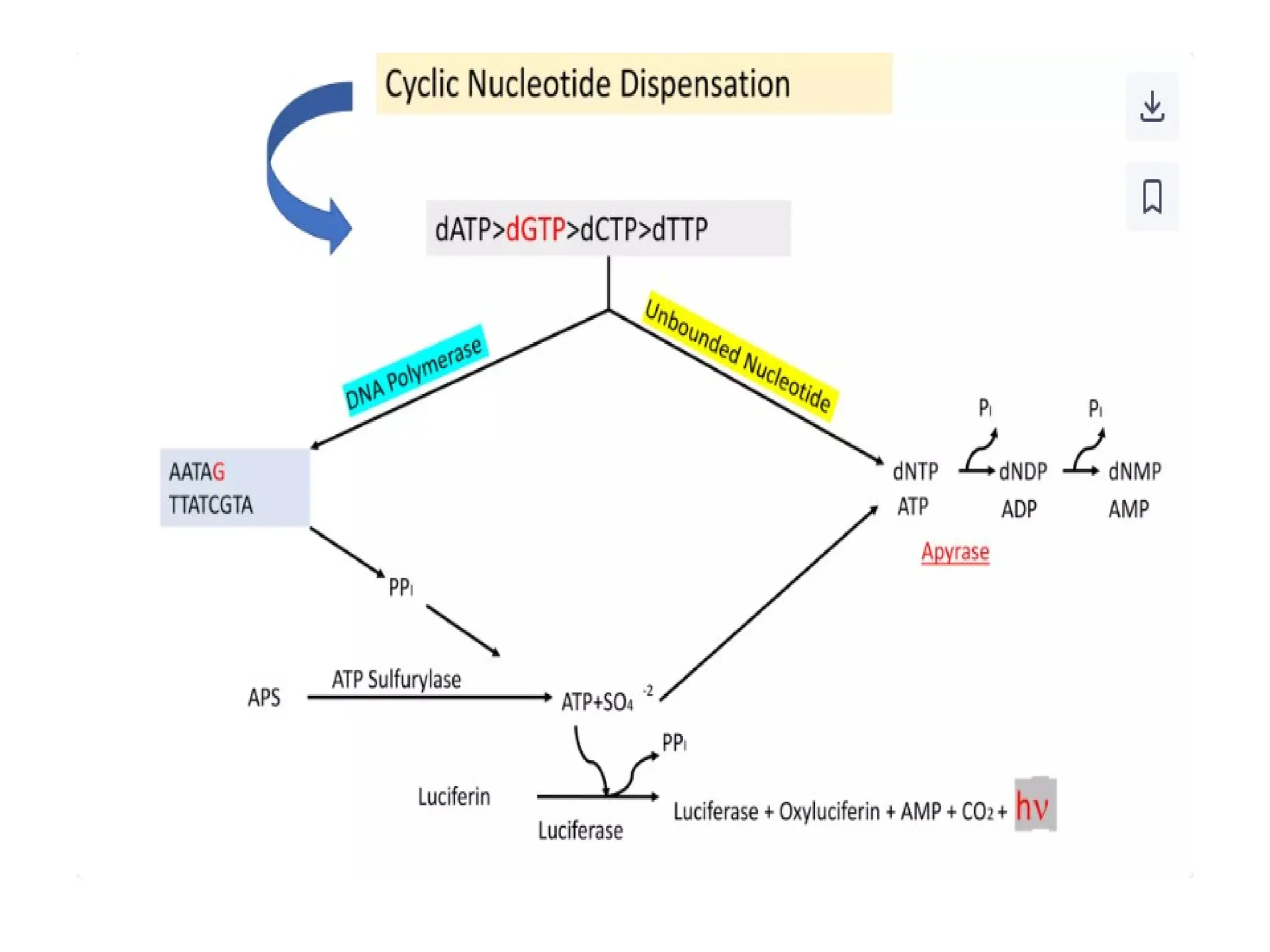
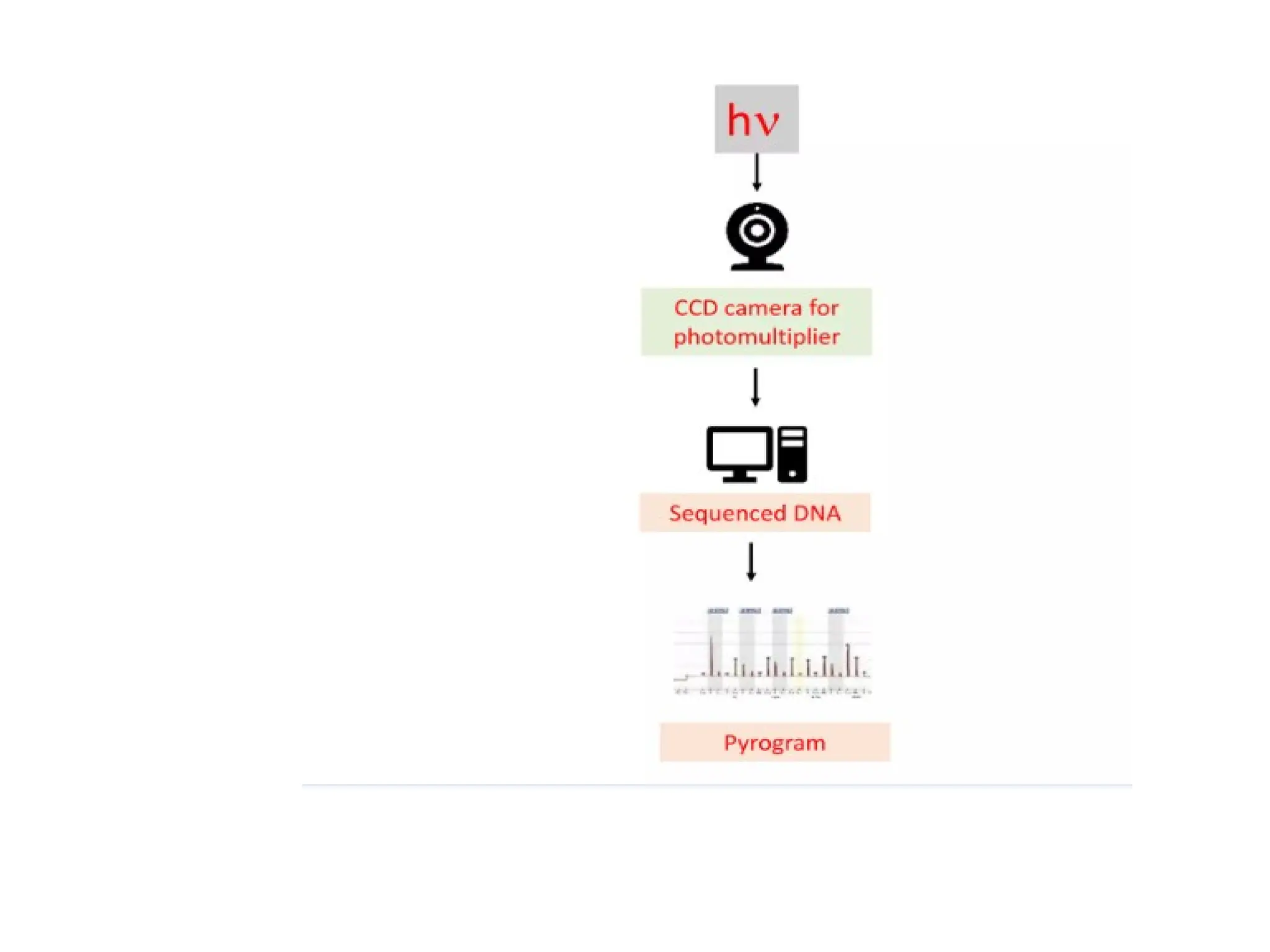
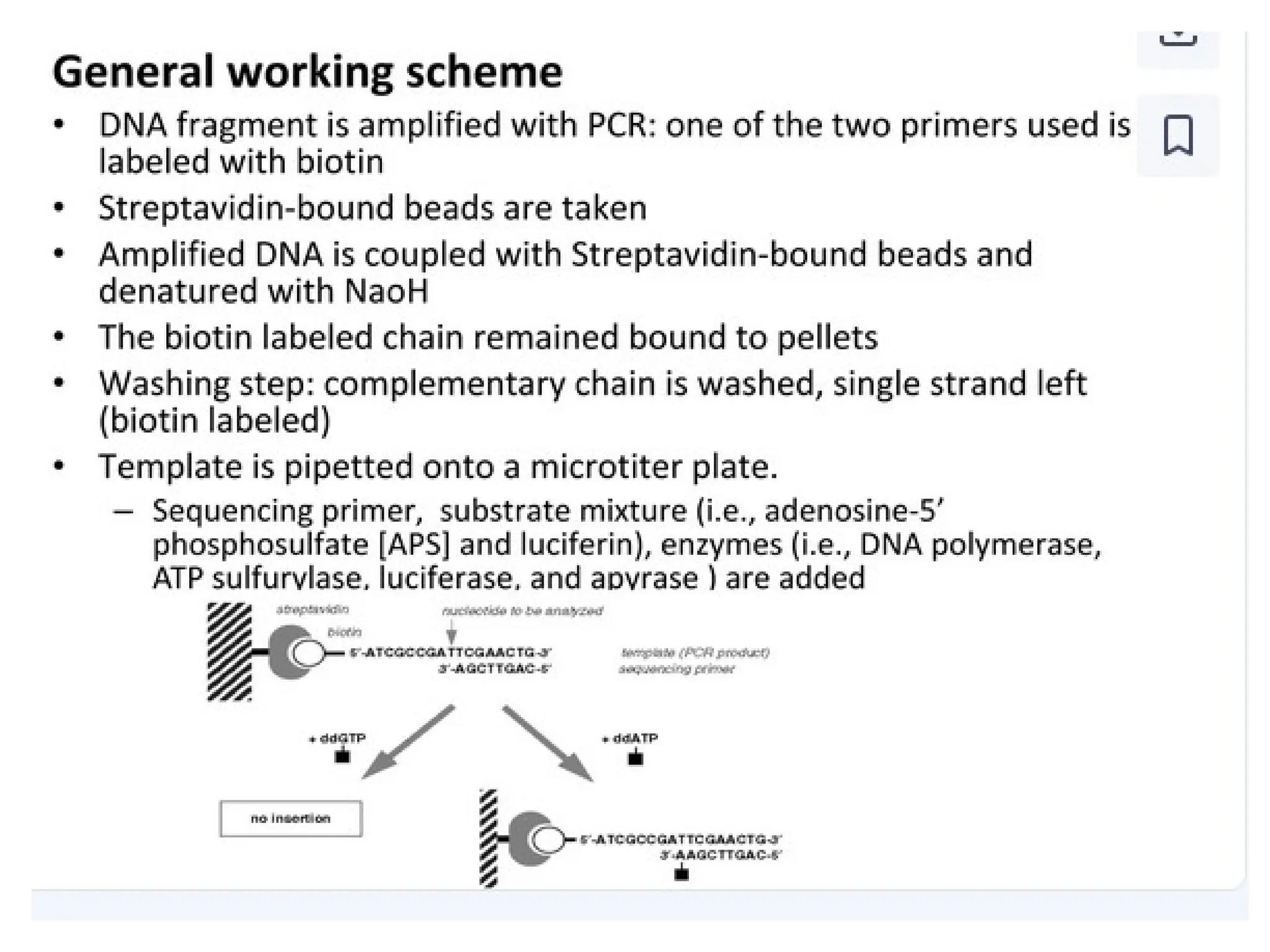
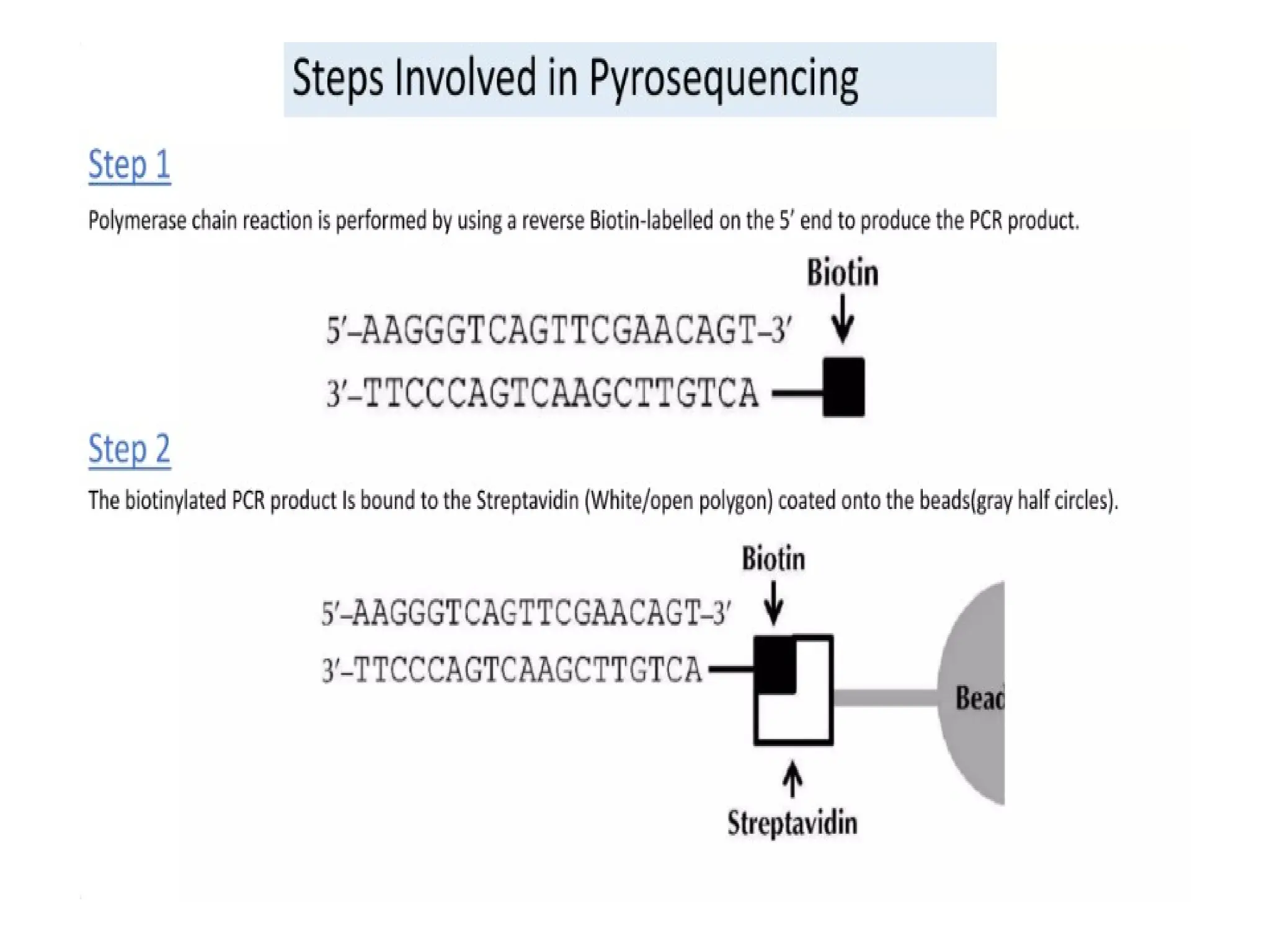
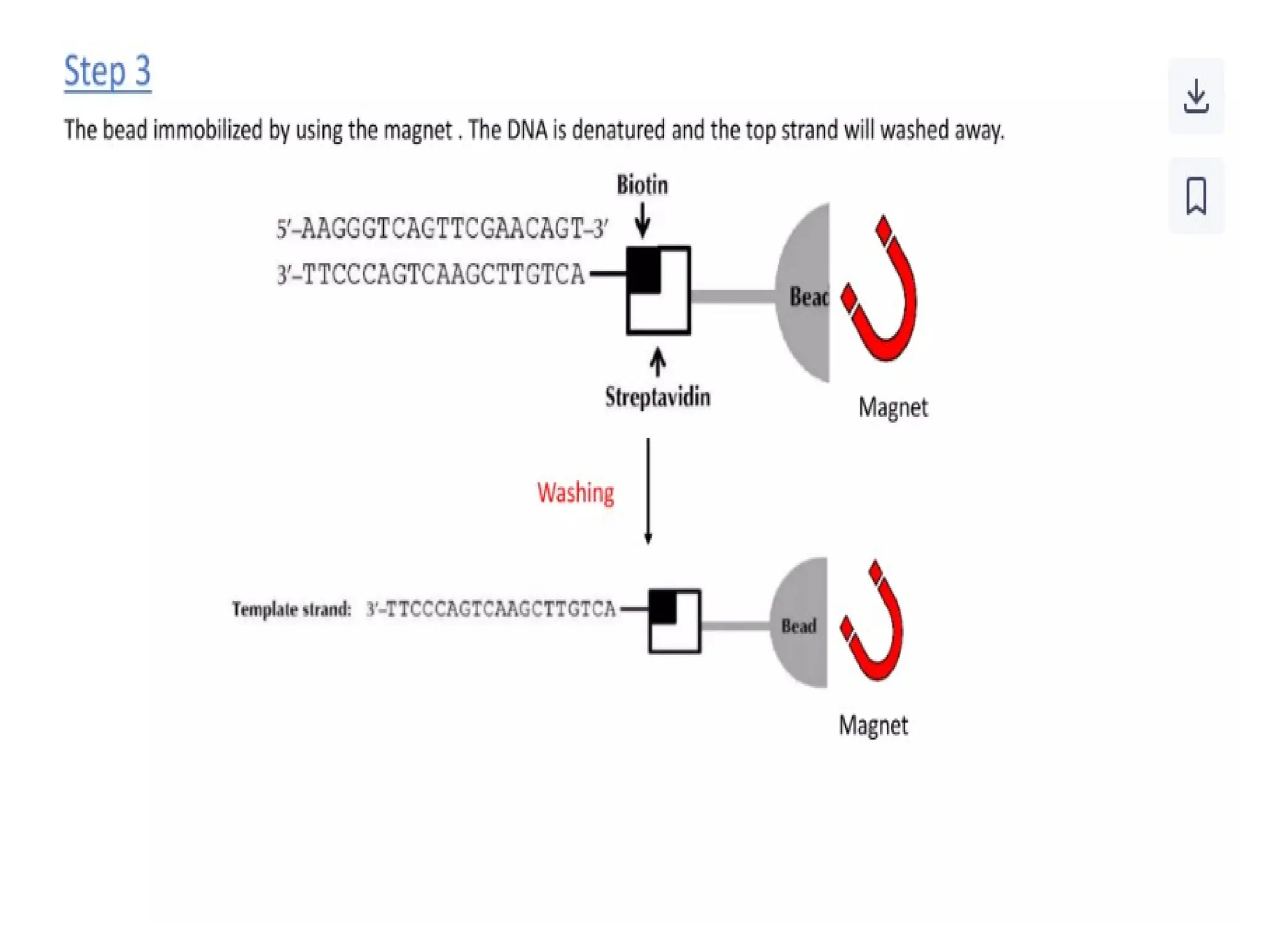
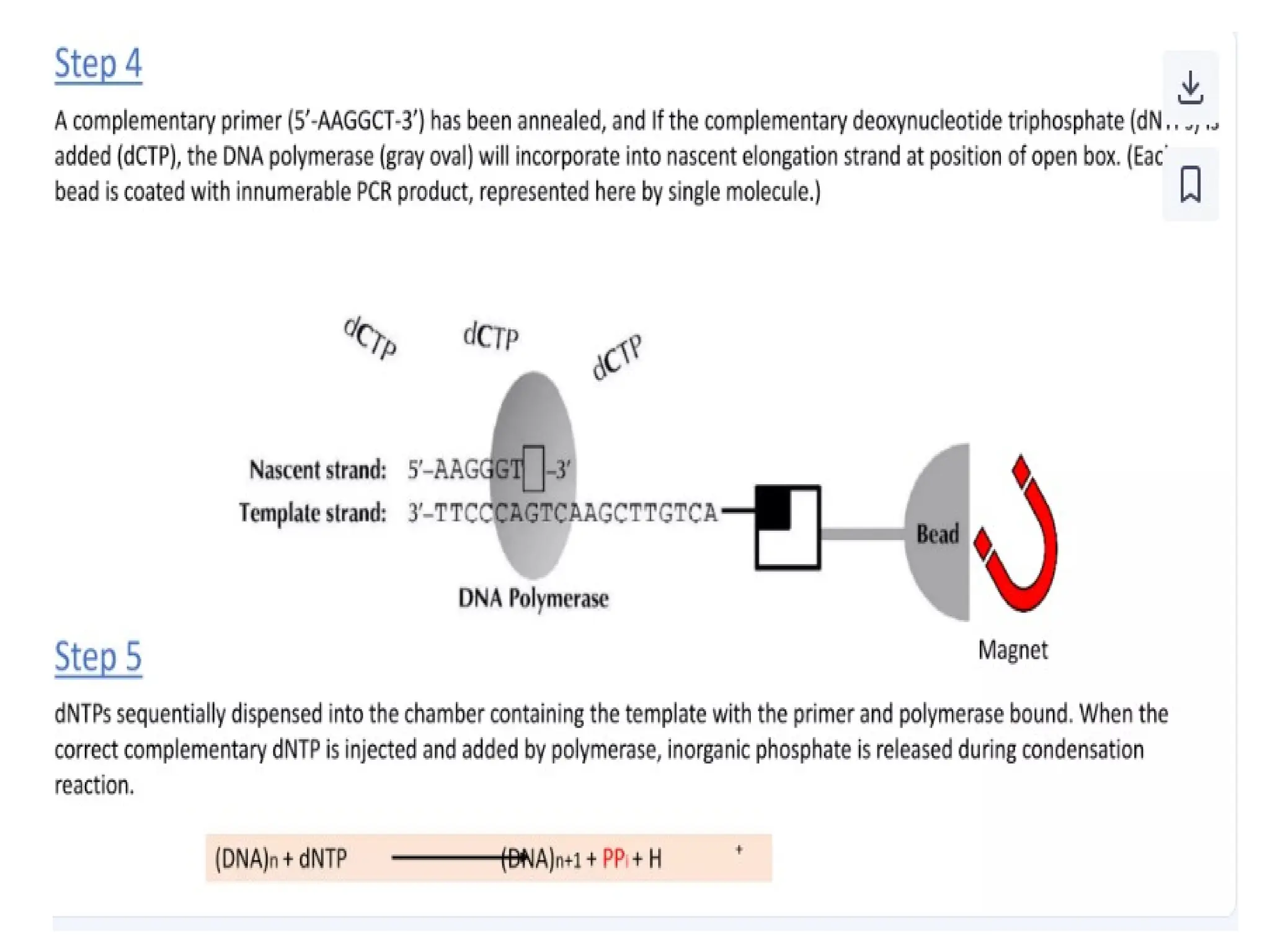
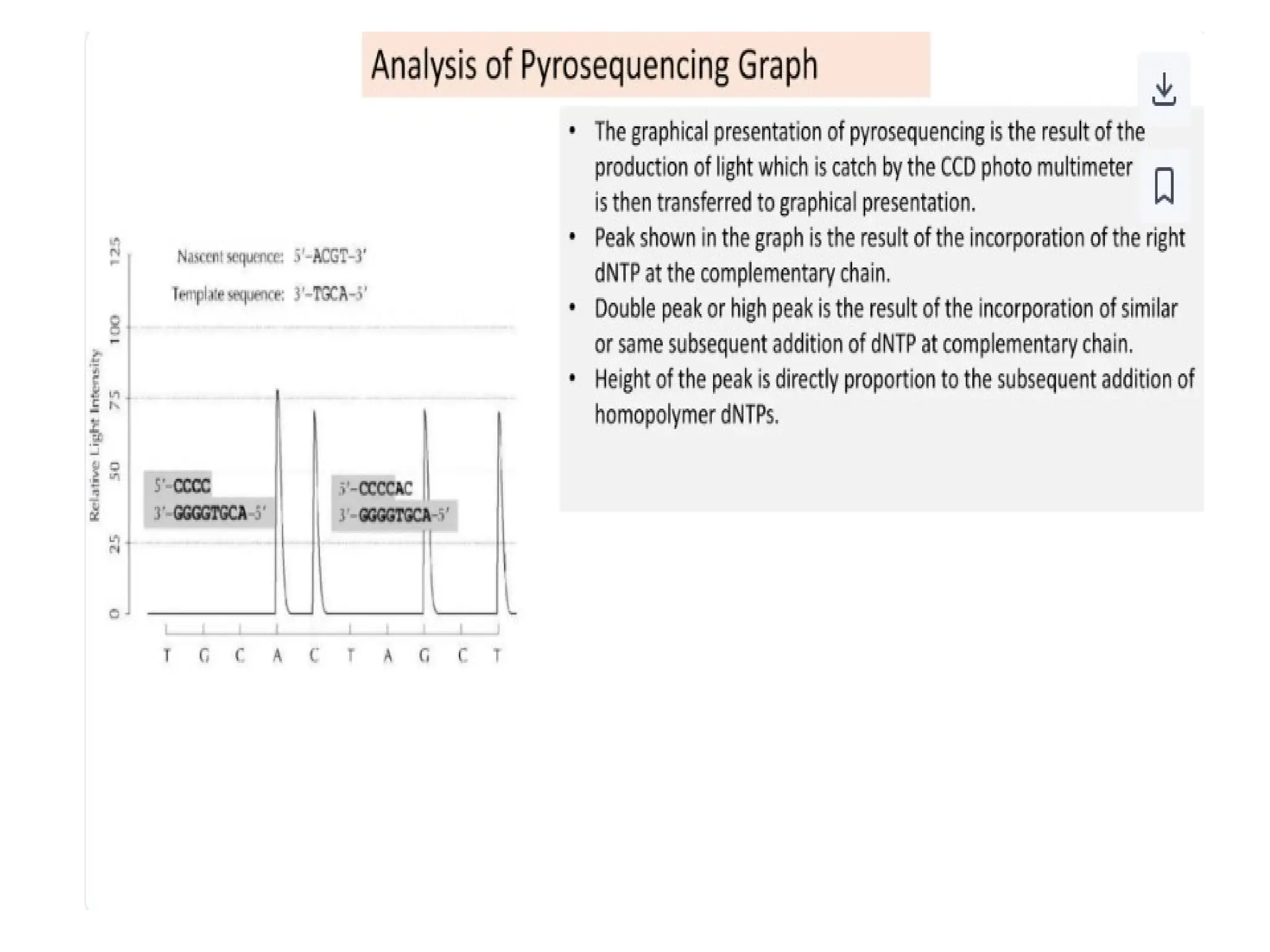
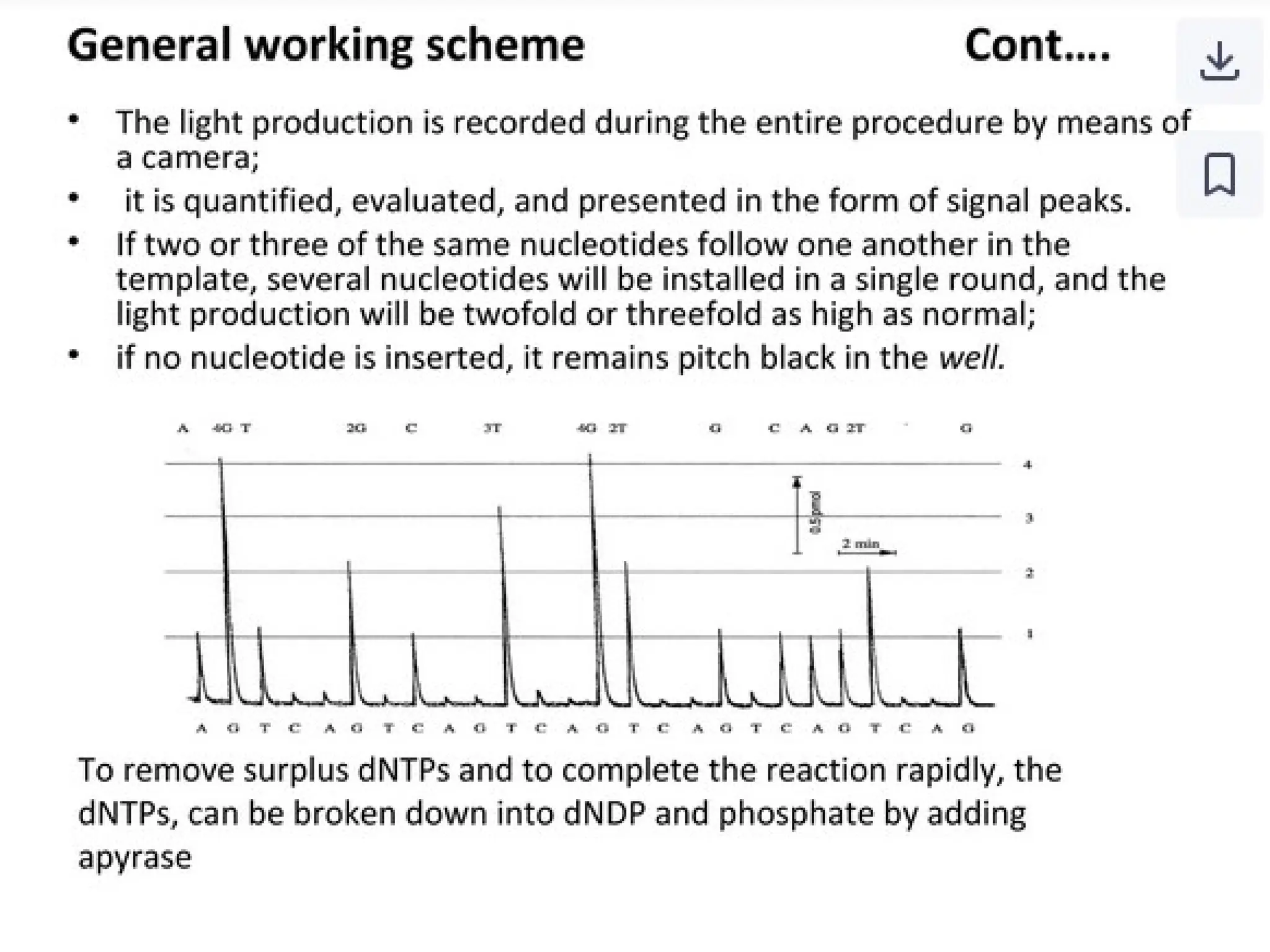
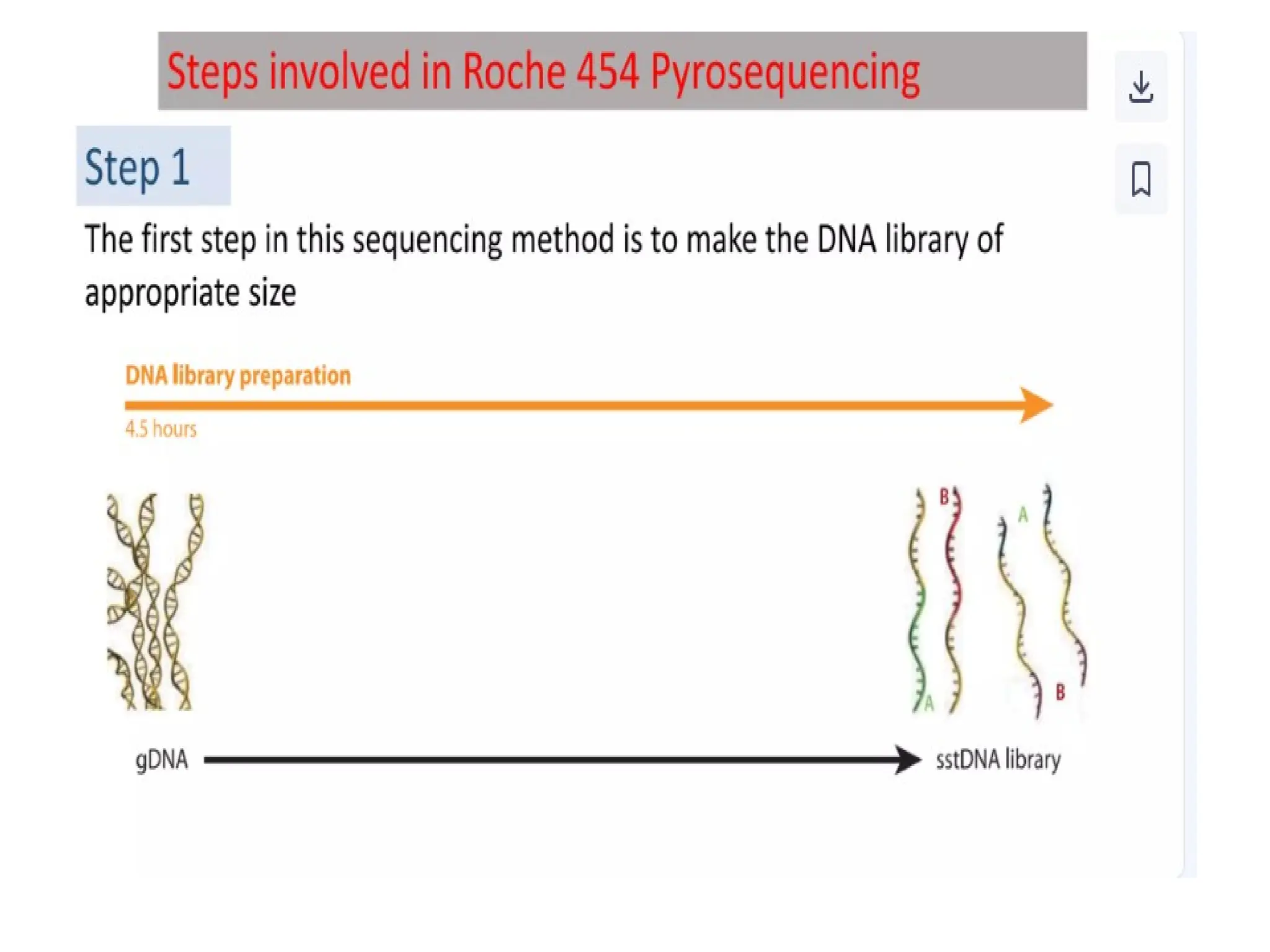
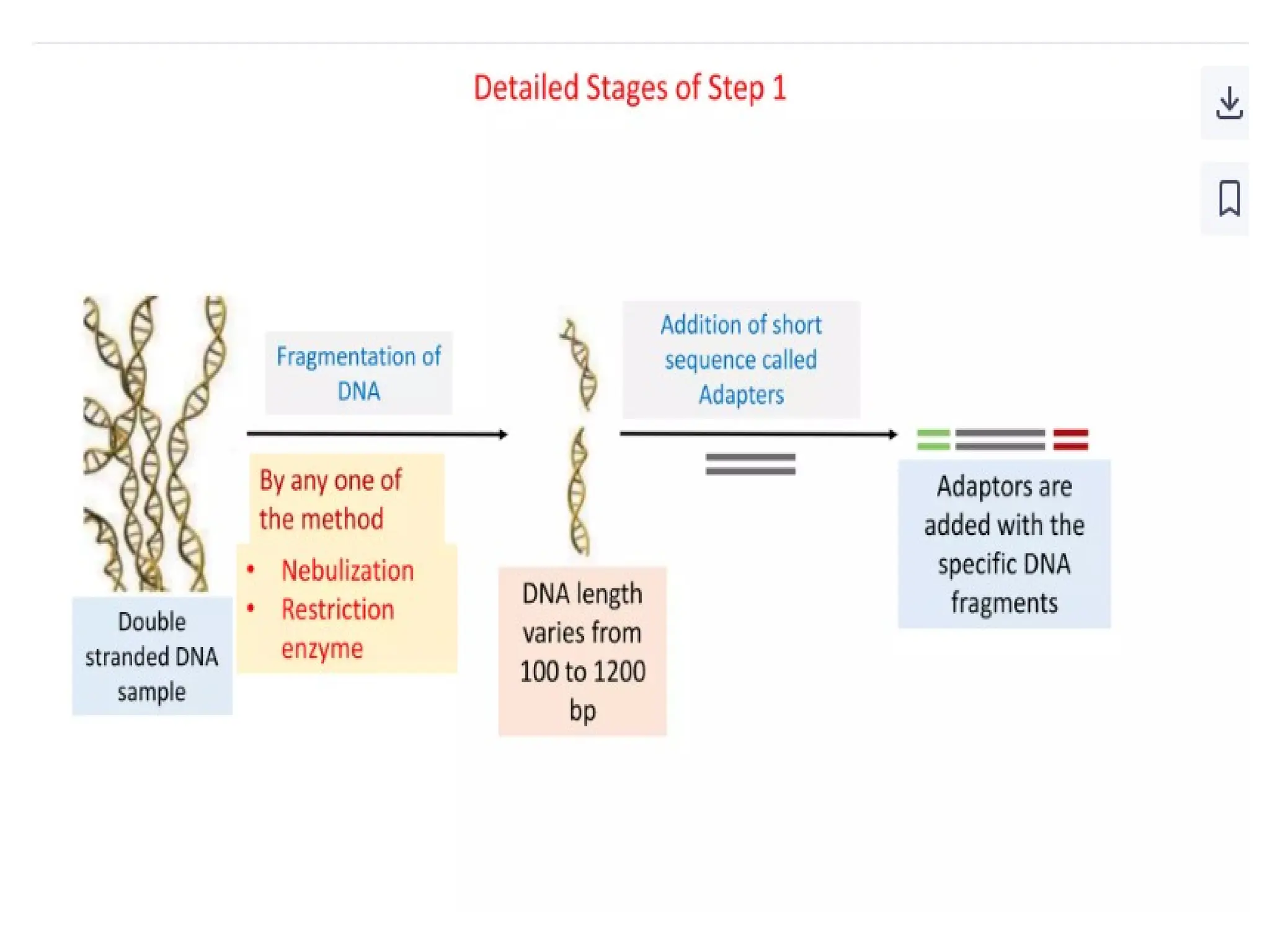
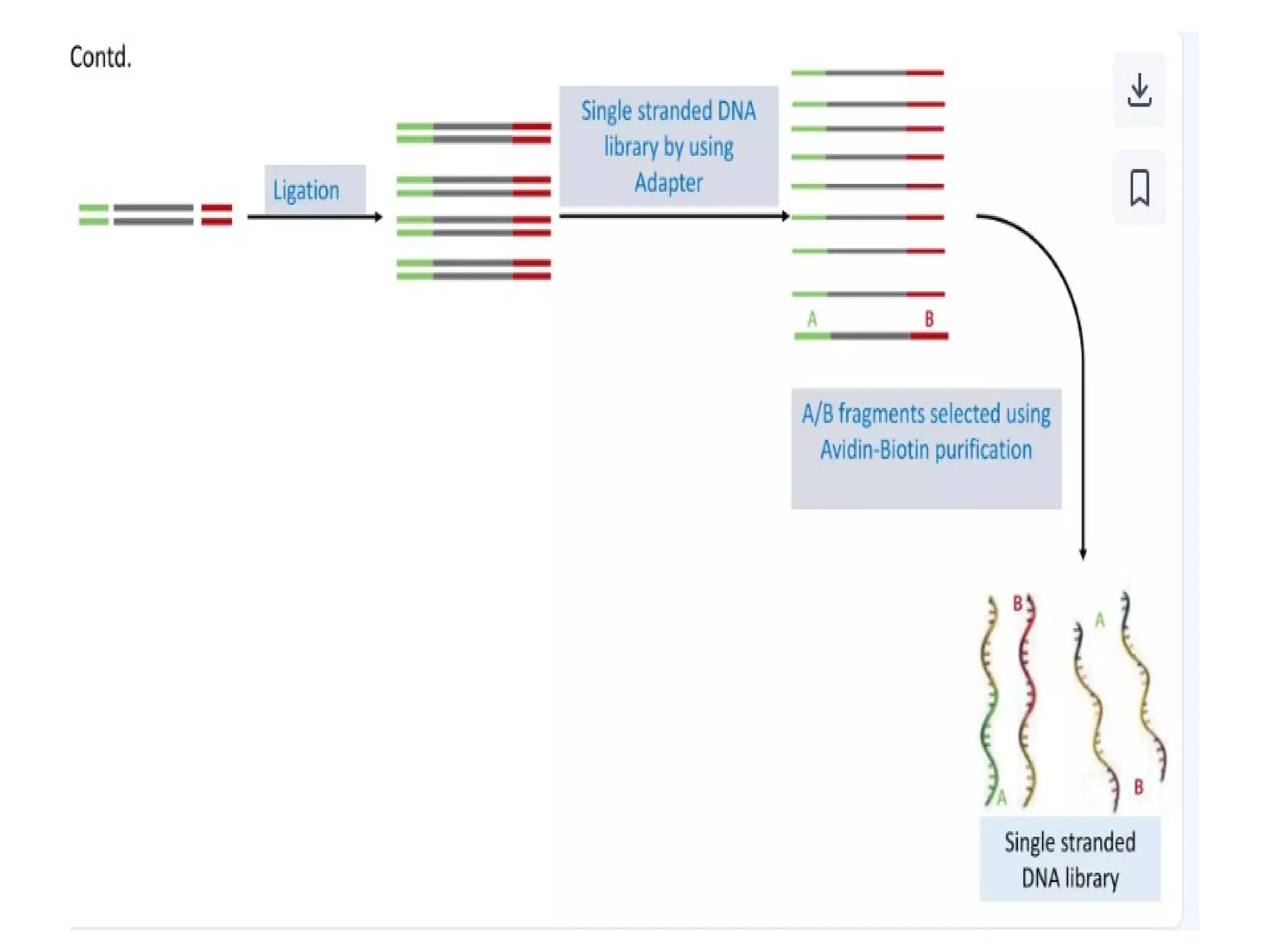
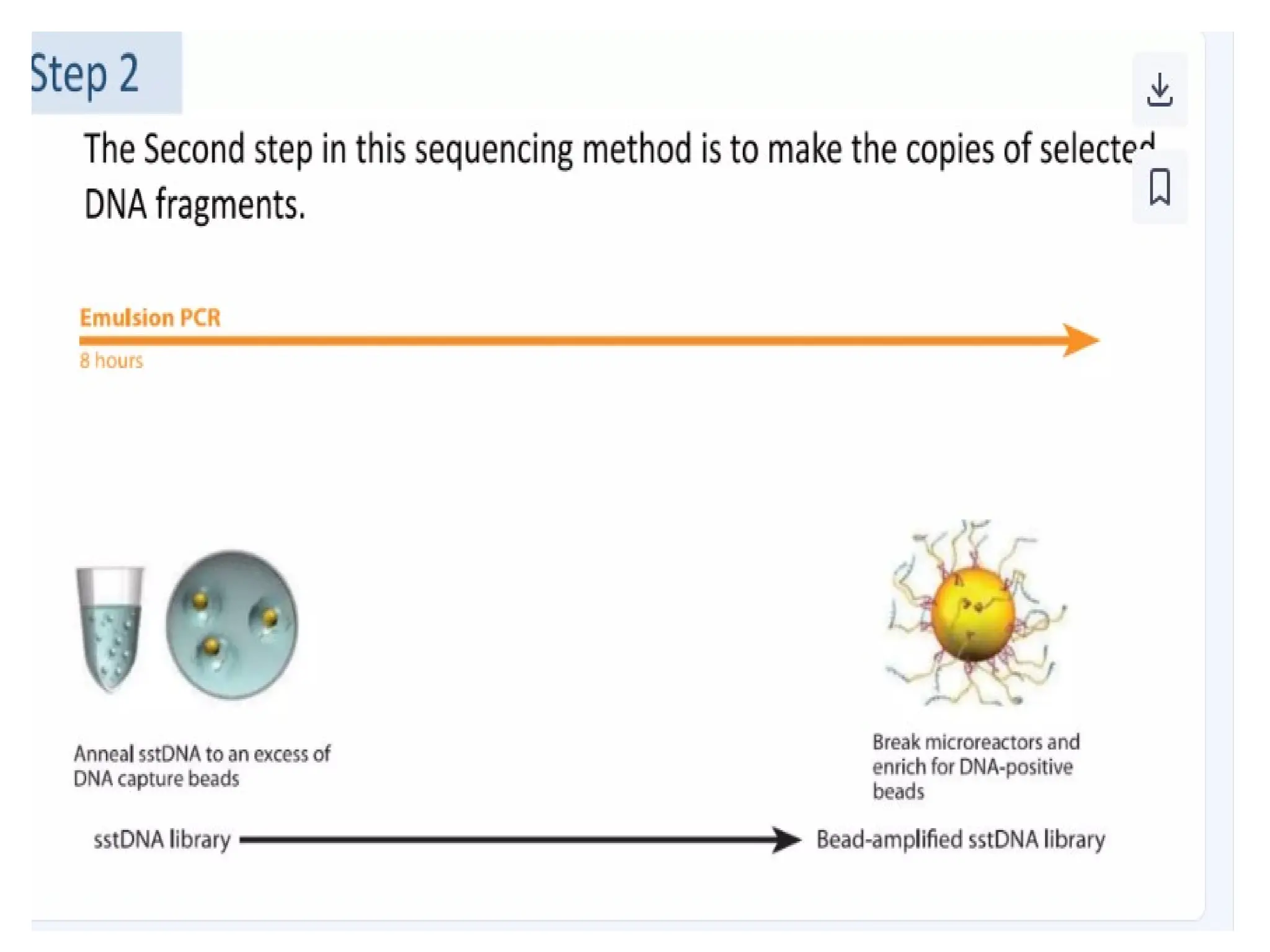
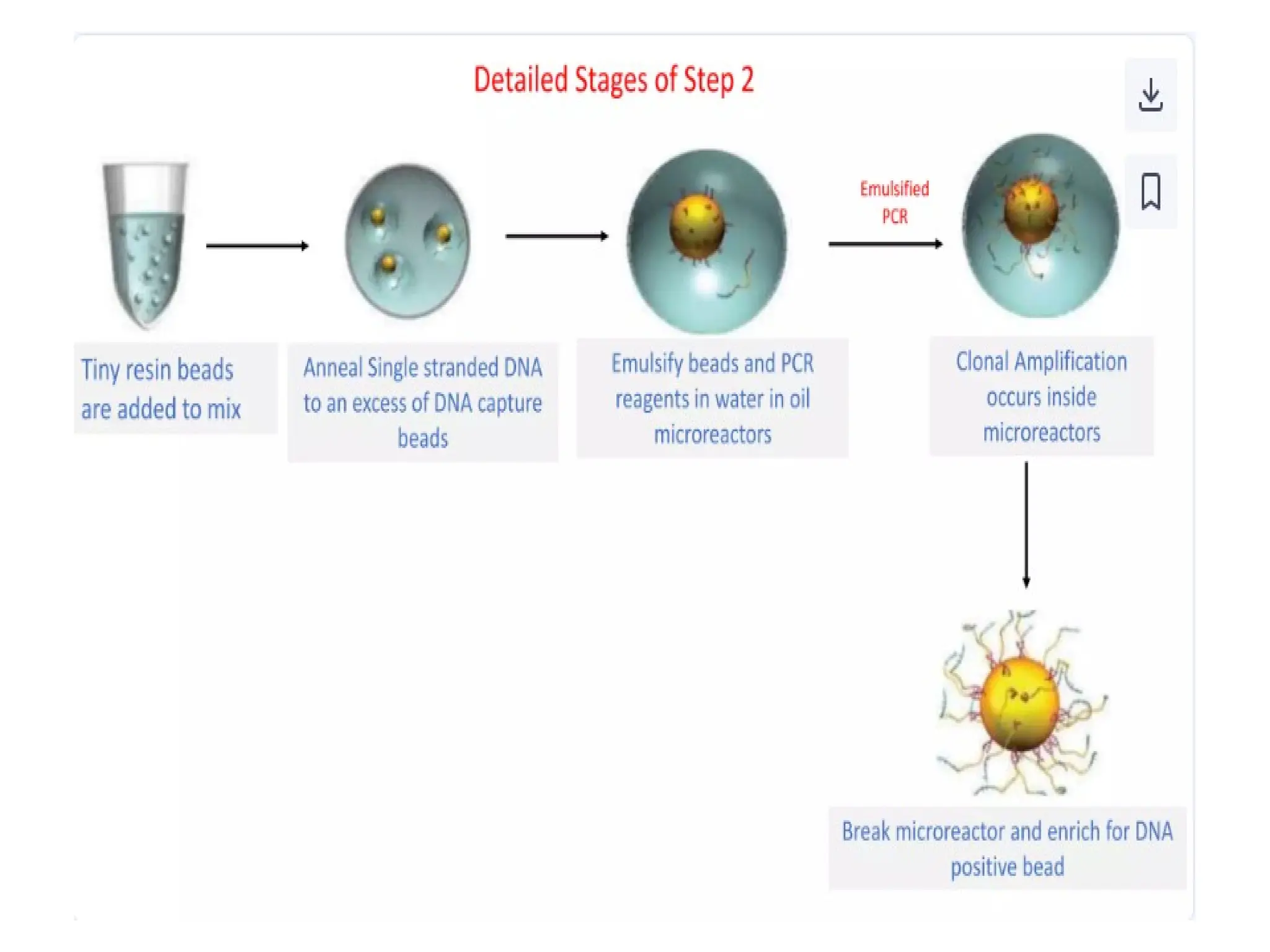
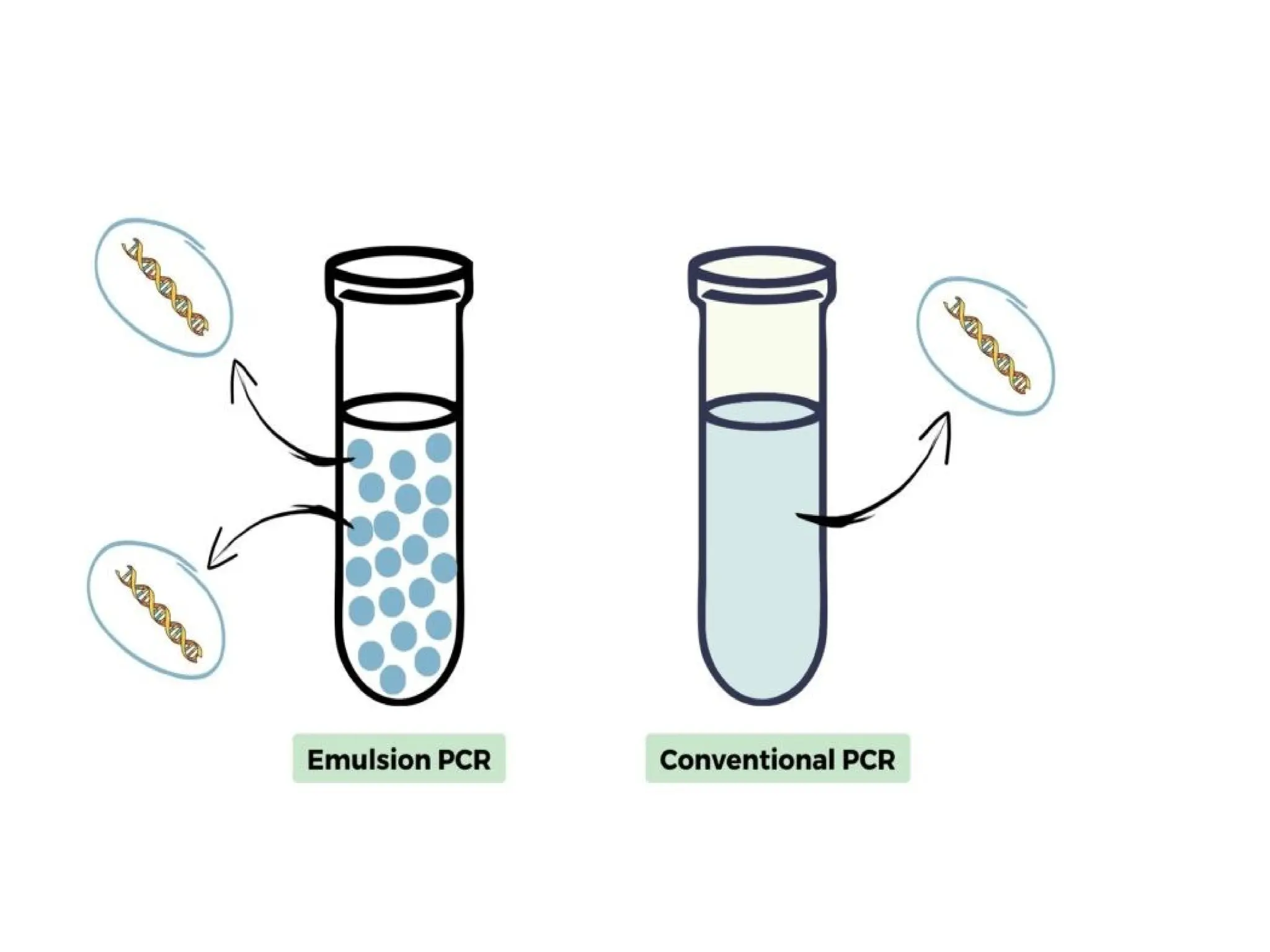
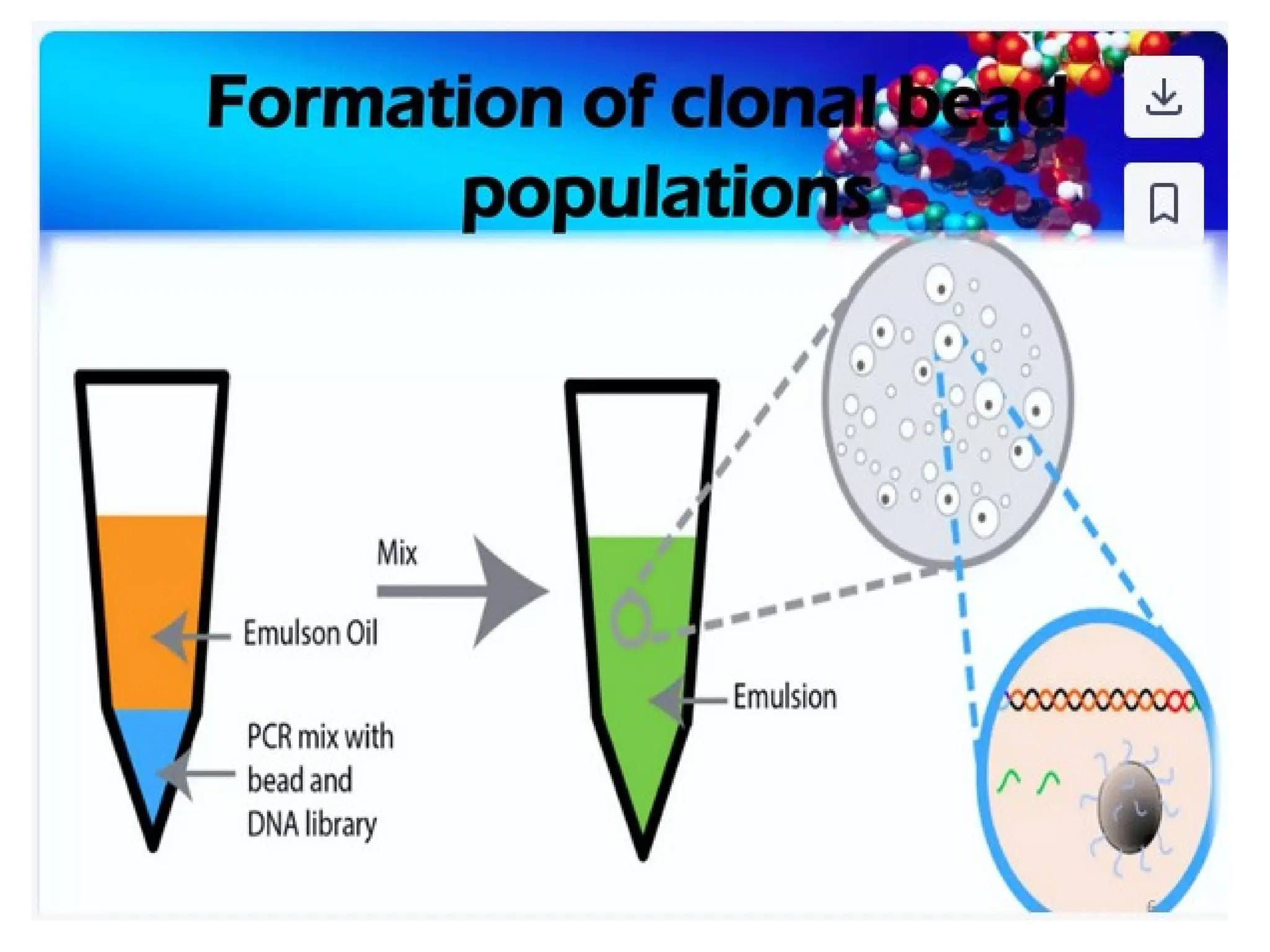
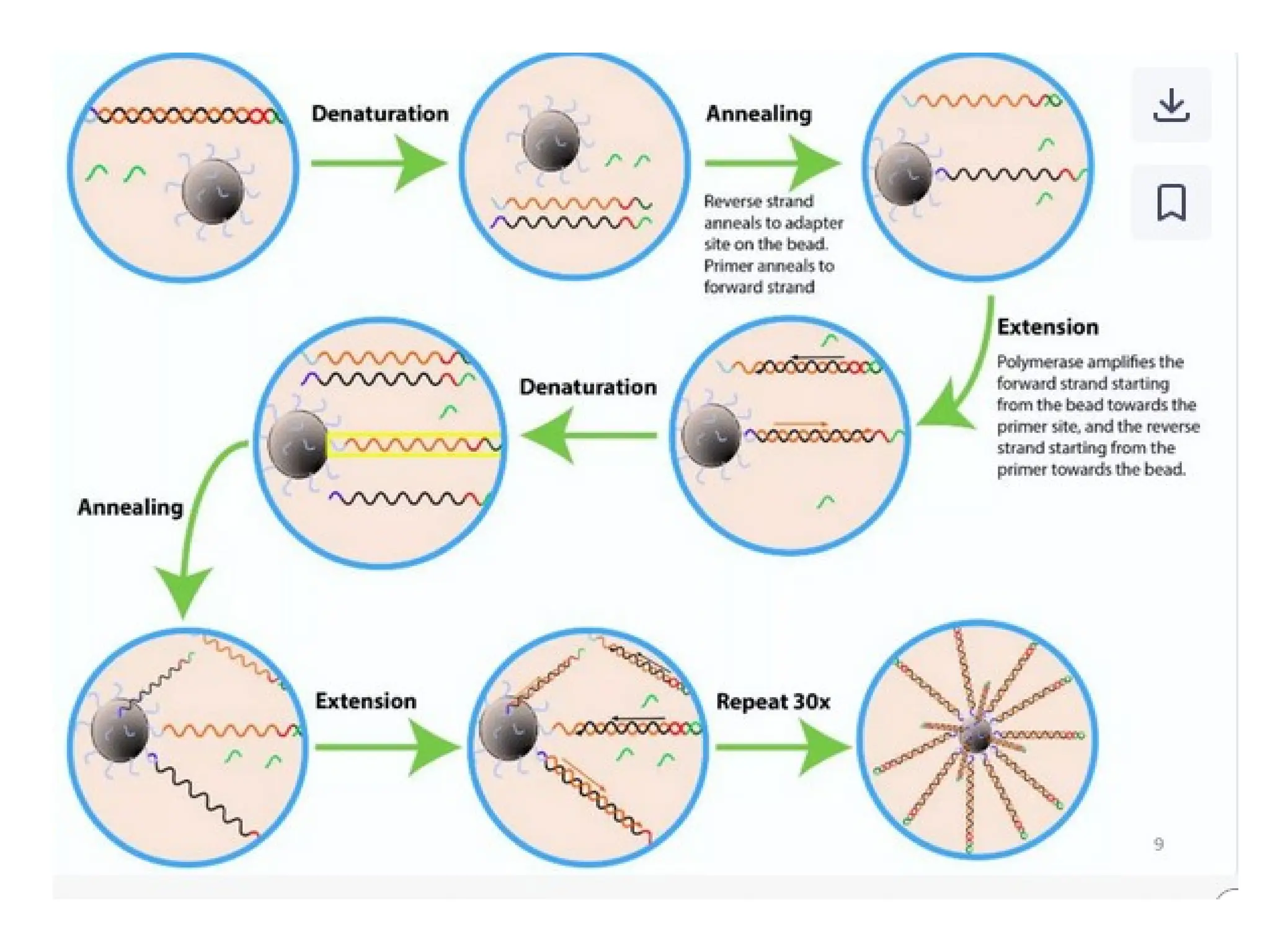
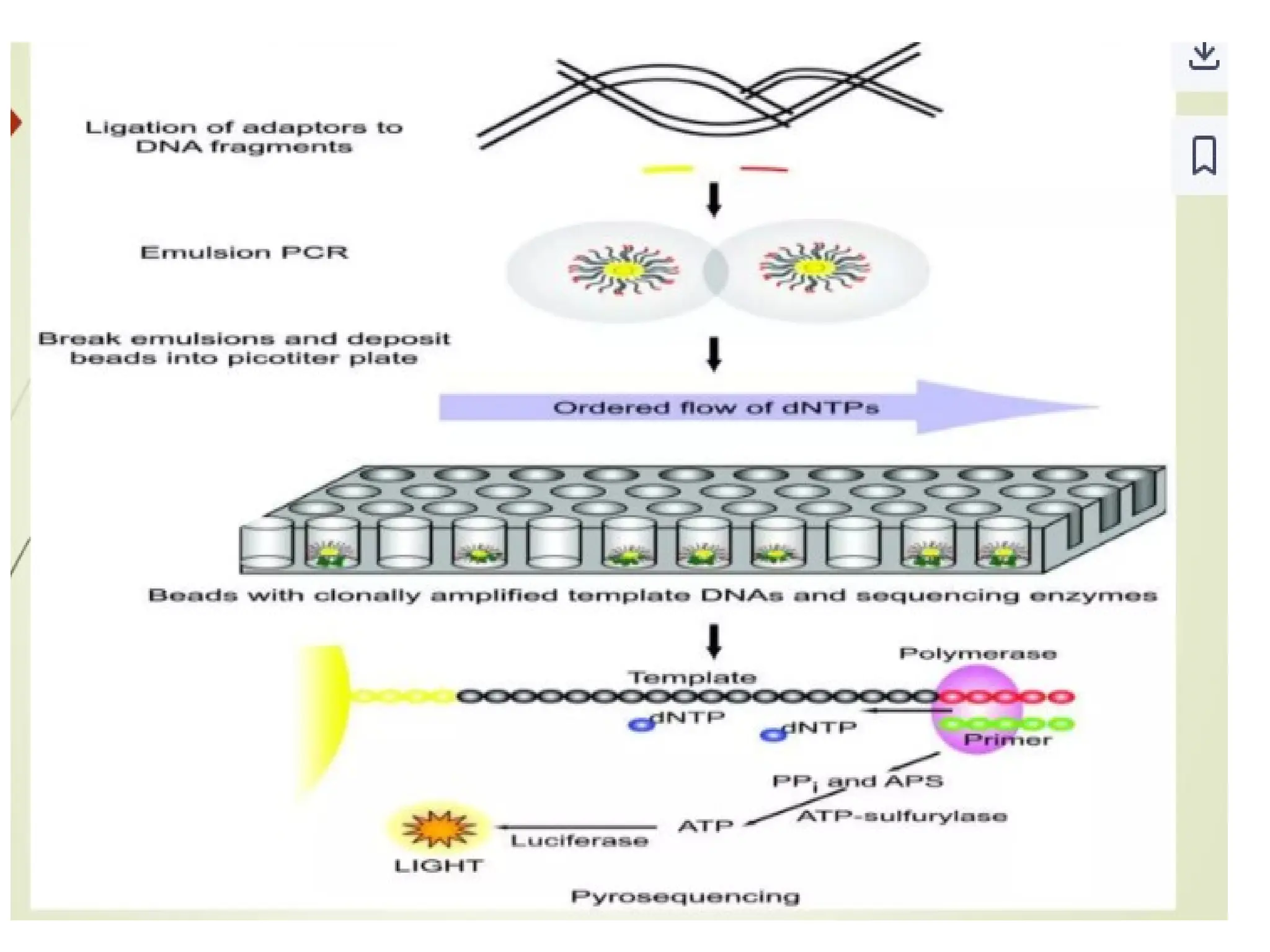
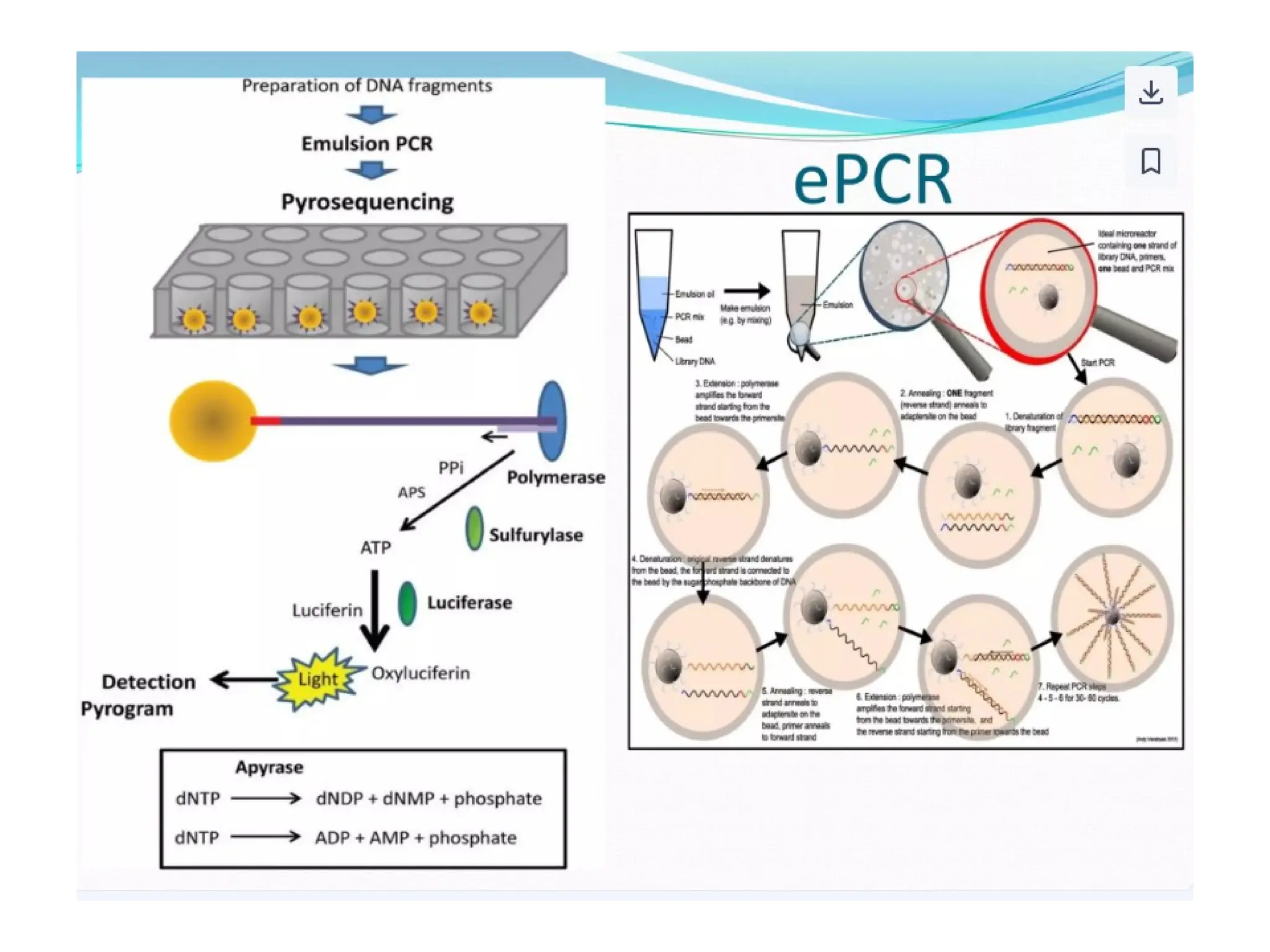
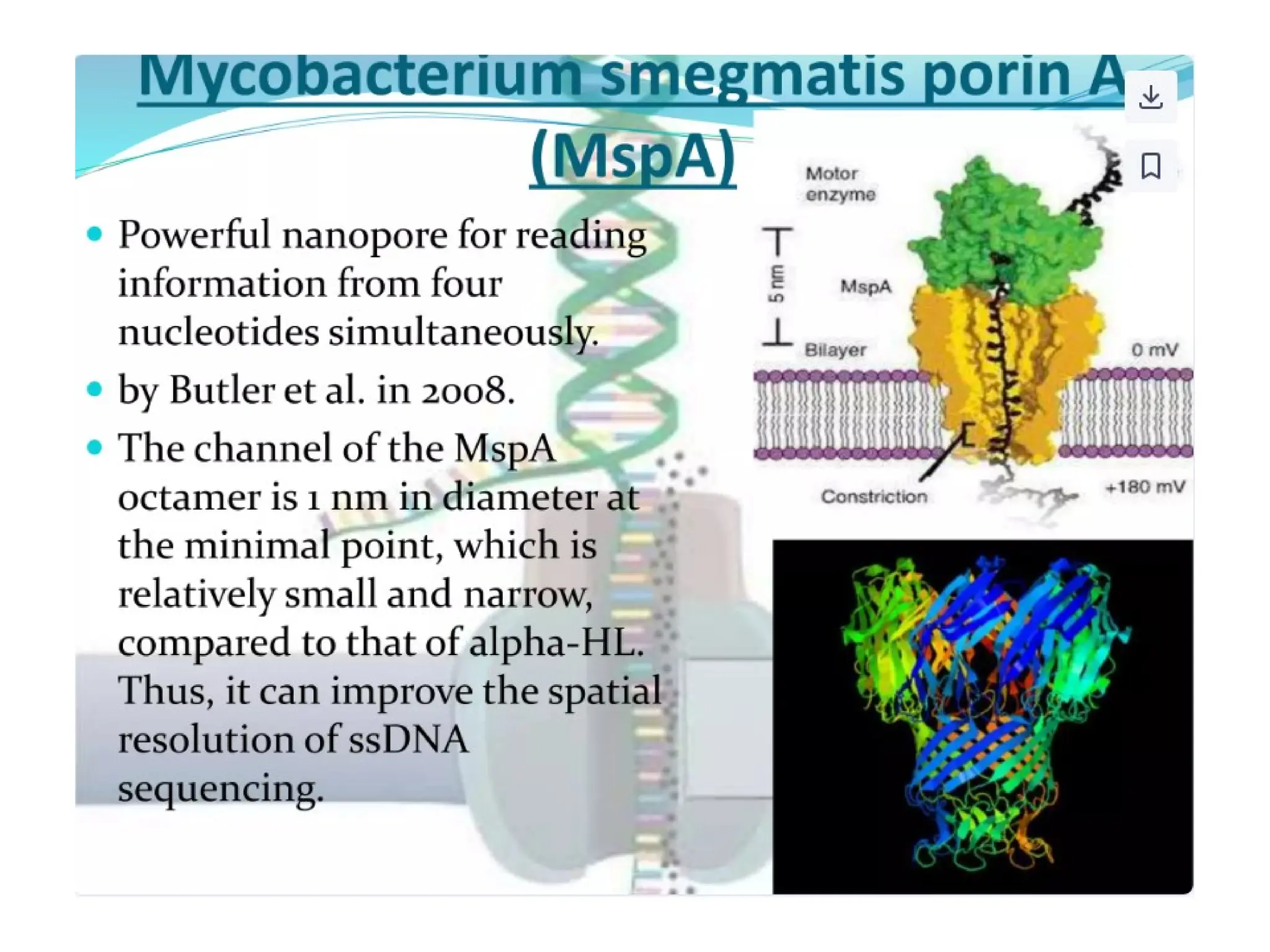
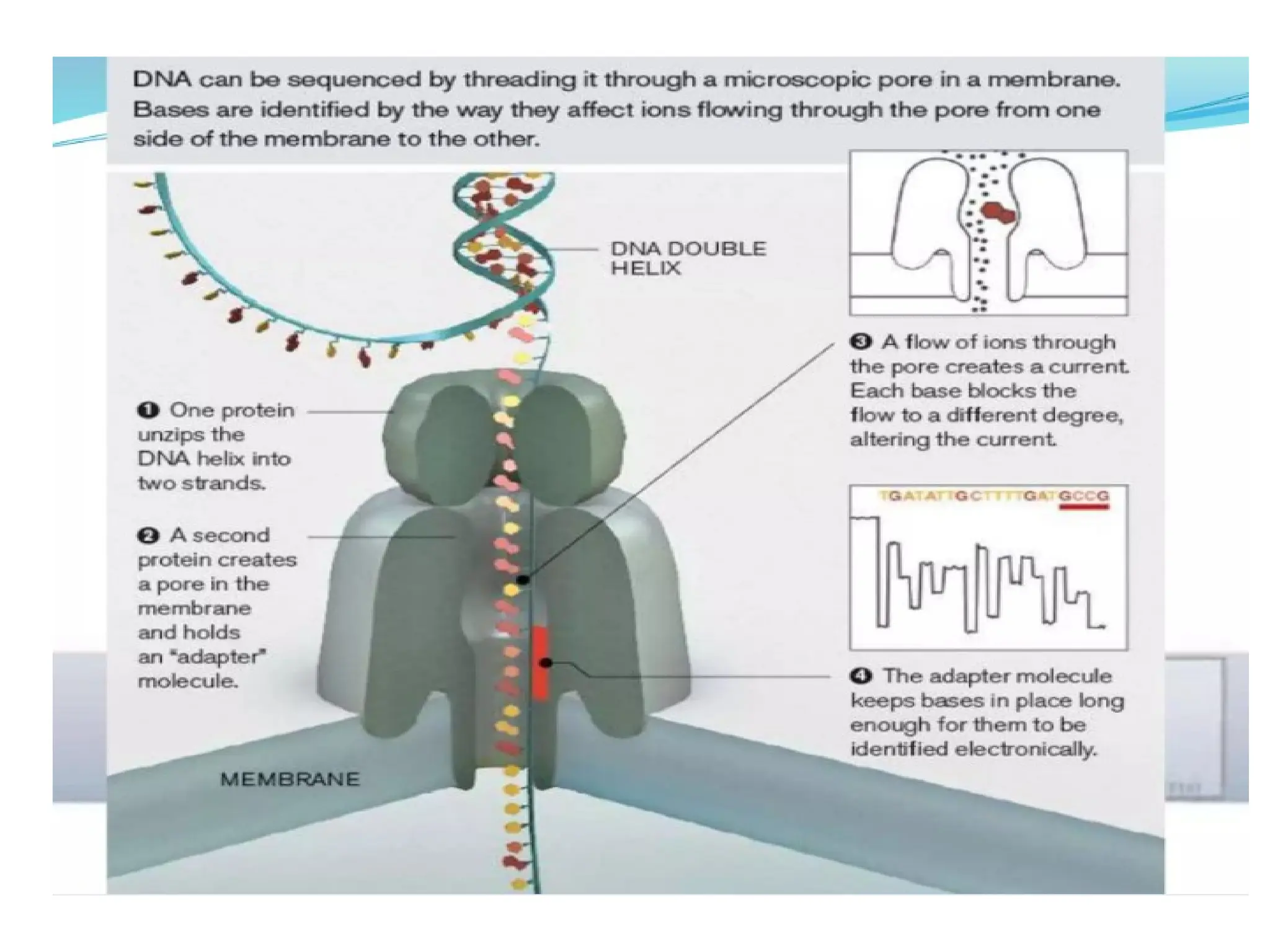
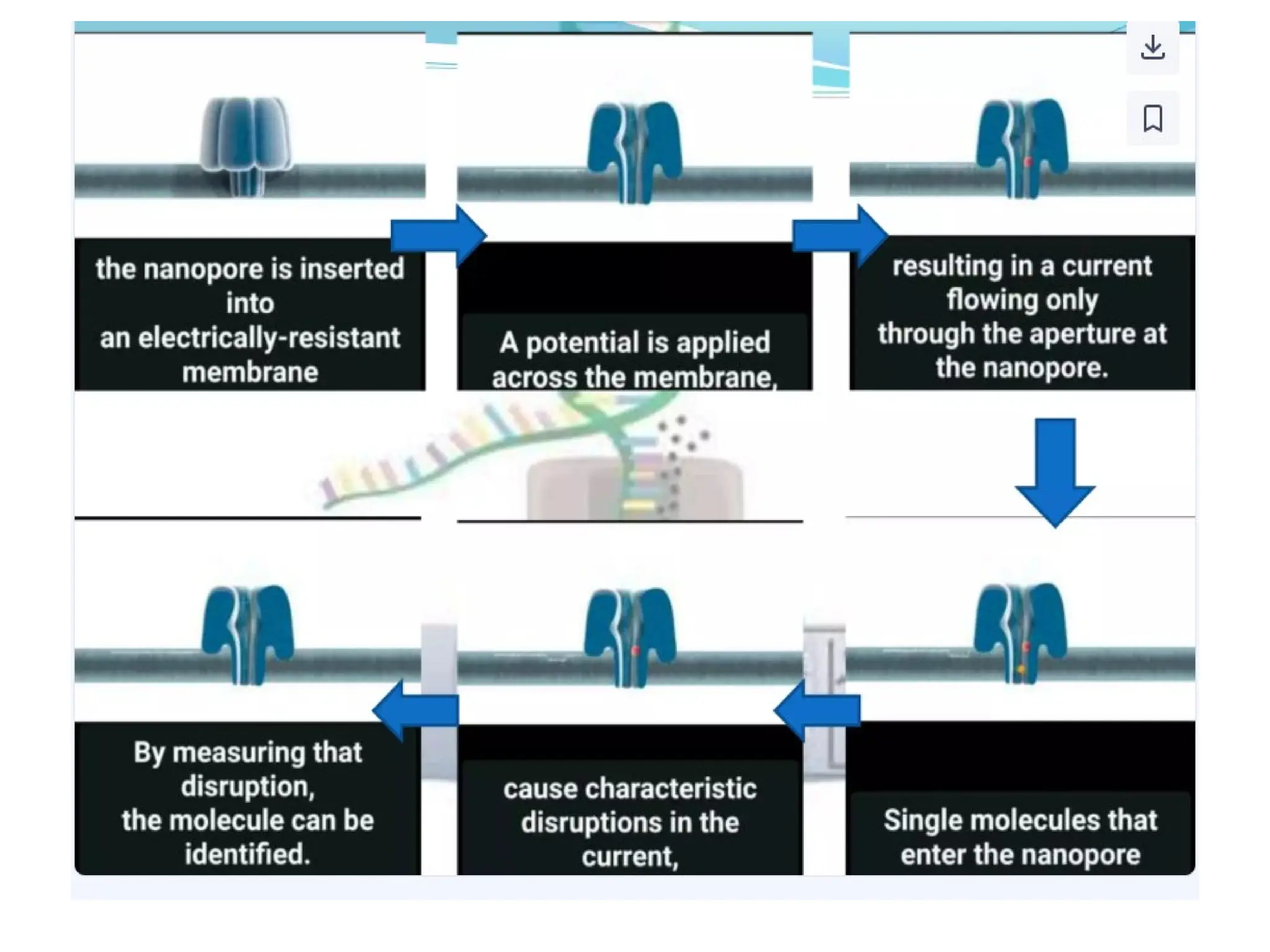
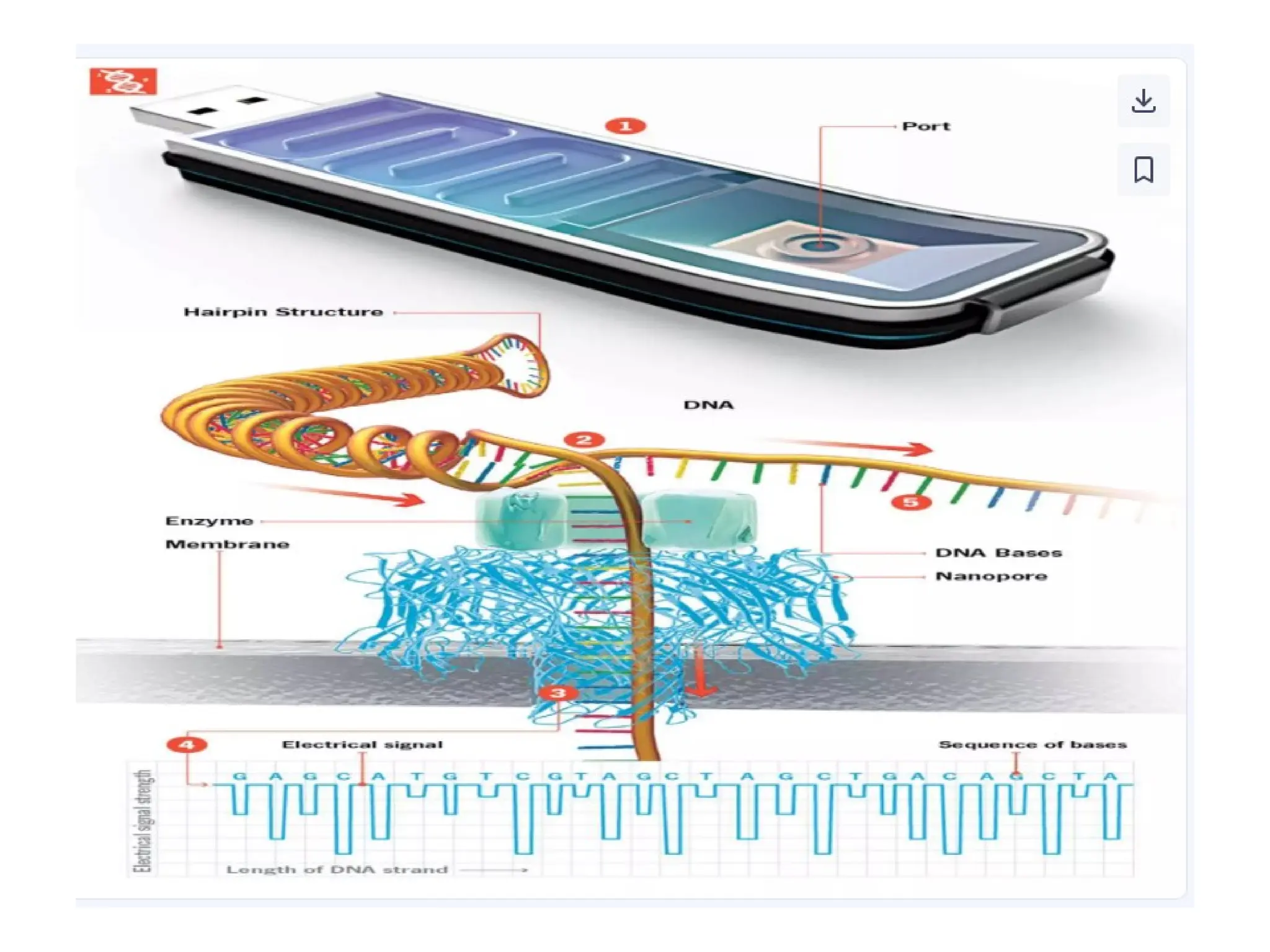
What is PCR?
•PCRis a technique that takes specific
sequence of DNA of small amount and
amplifies it to be used for further testing.
•In vitro technique
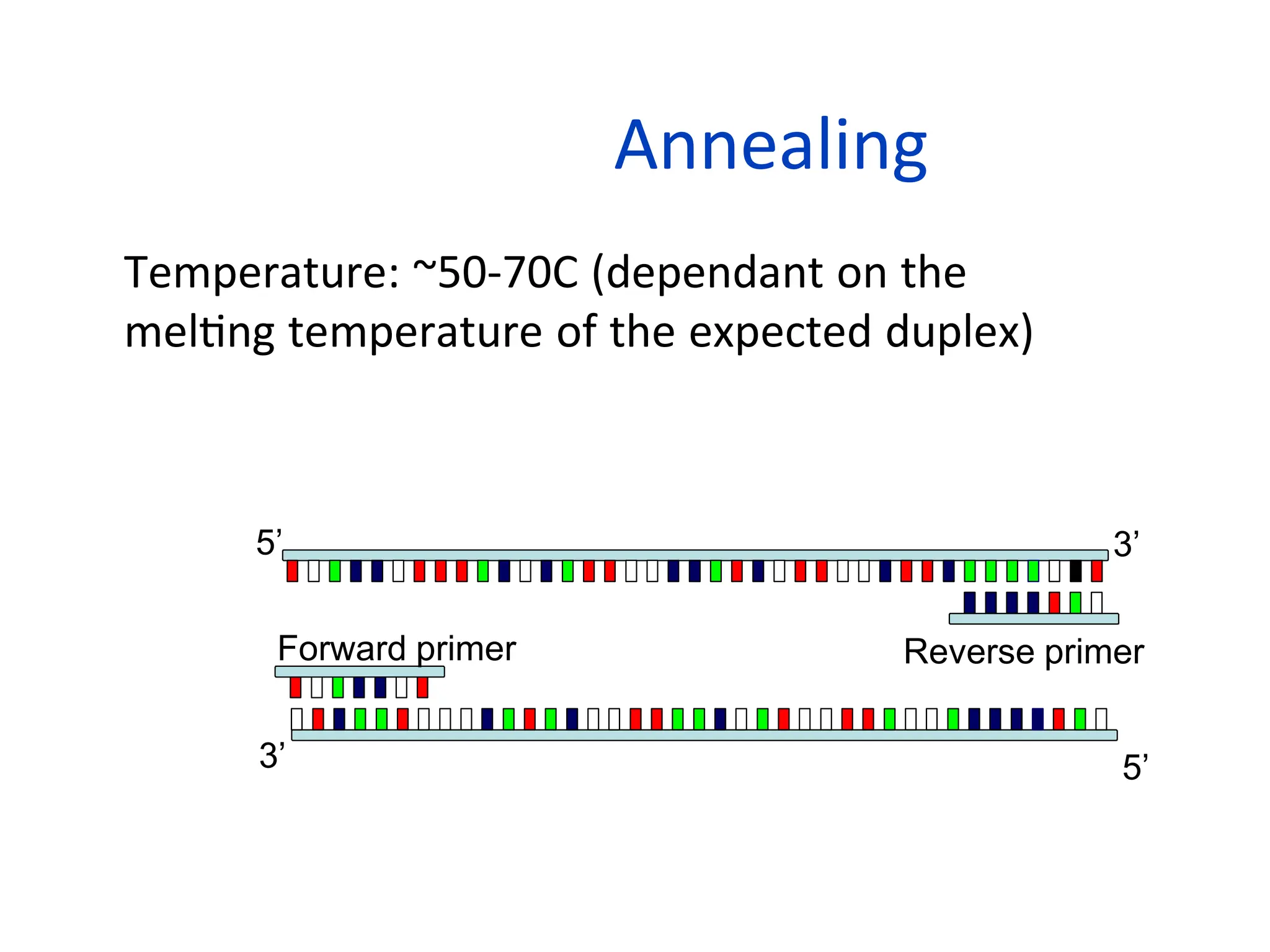
Annealing
• Temperature: ~50-70C(dependant on the
melting temperature of the expected duplex)
• Primers bind to their complementary sequences
5’
3’
5’ 3’
Forward primer Reverse primer
Applications of PCR
MolecularIdentification
Molecular Archaeology
Molecular Epidemiology
Sequencing
Bioinformatics Genomic
Cloning
Genetic Engineering
Site-directed mutagenesis
Gene Expression Studies
Molecular Ecology DNA
fingerprinting
Classification of organisms
Human Genome Project
Genotyping
Pre-natal diagnosis
Mutation screening Drug
discovery Genetic matching
Detection of pathogens
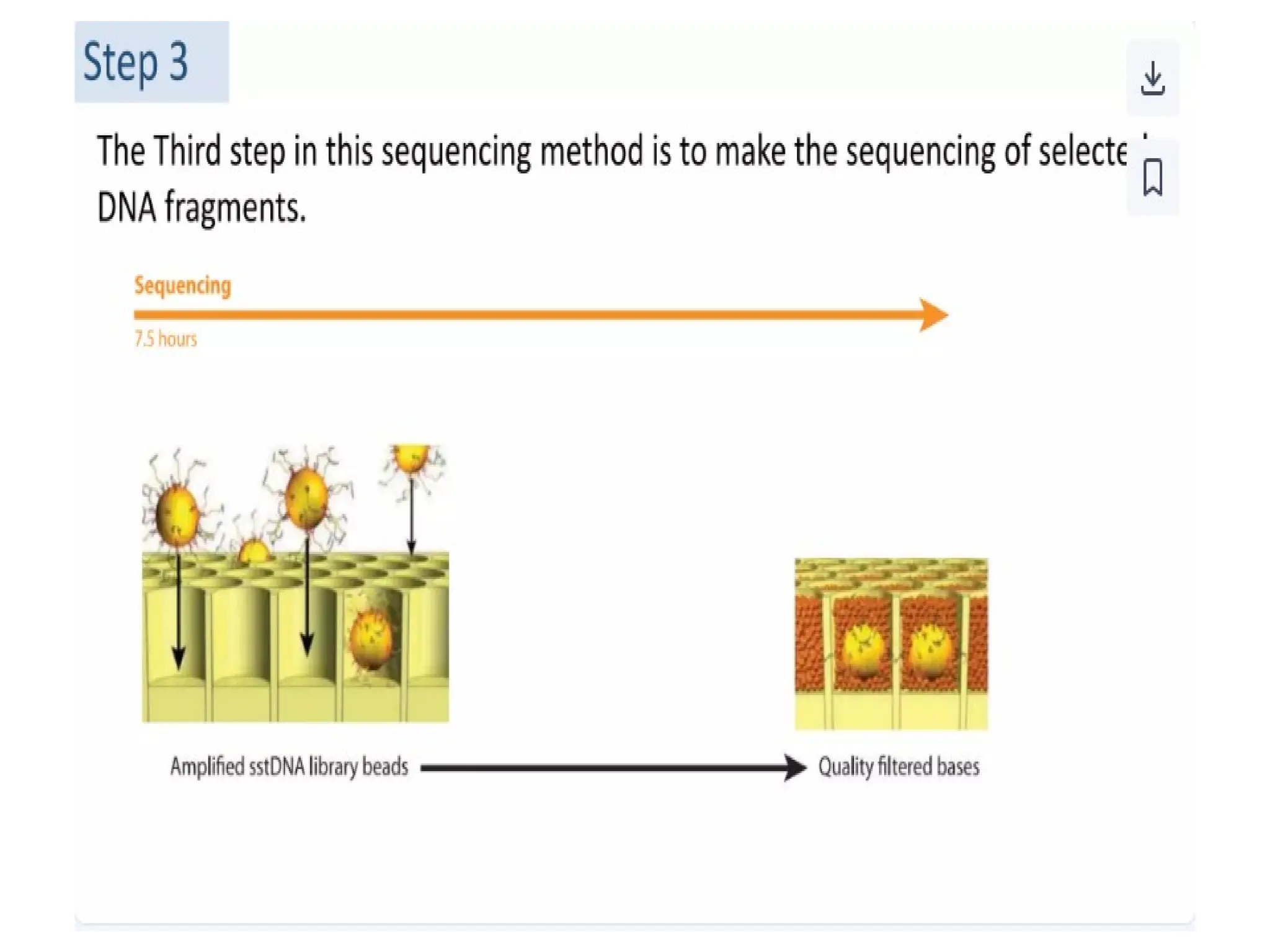
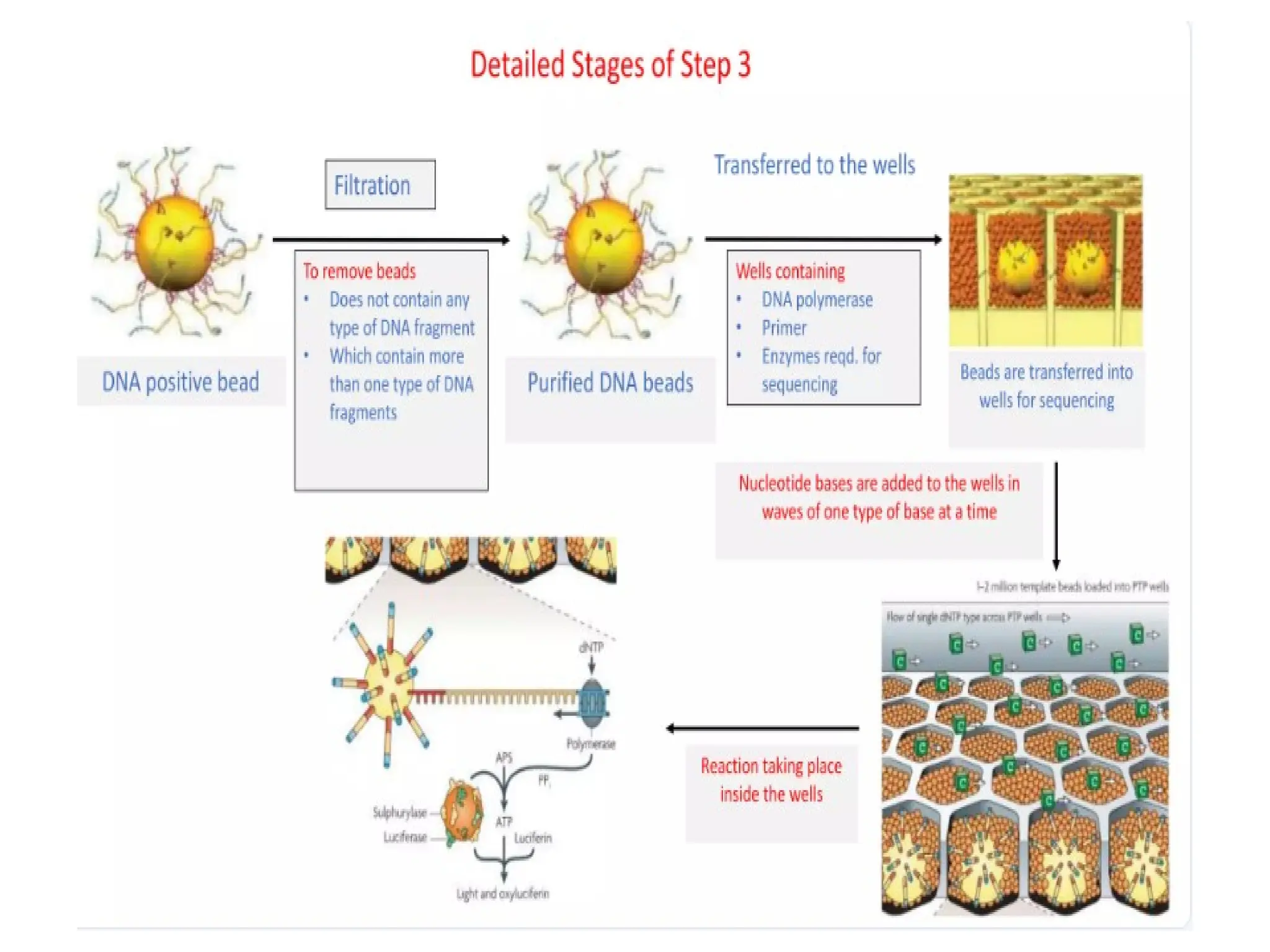
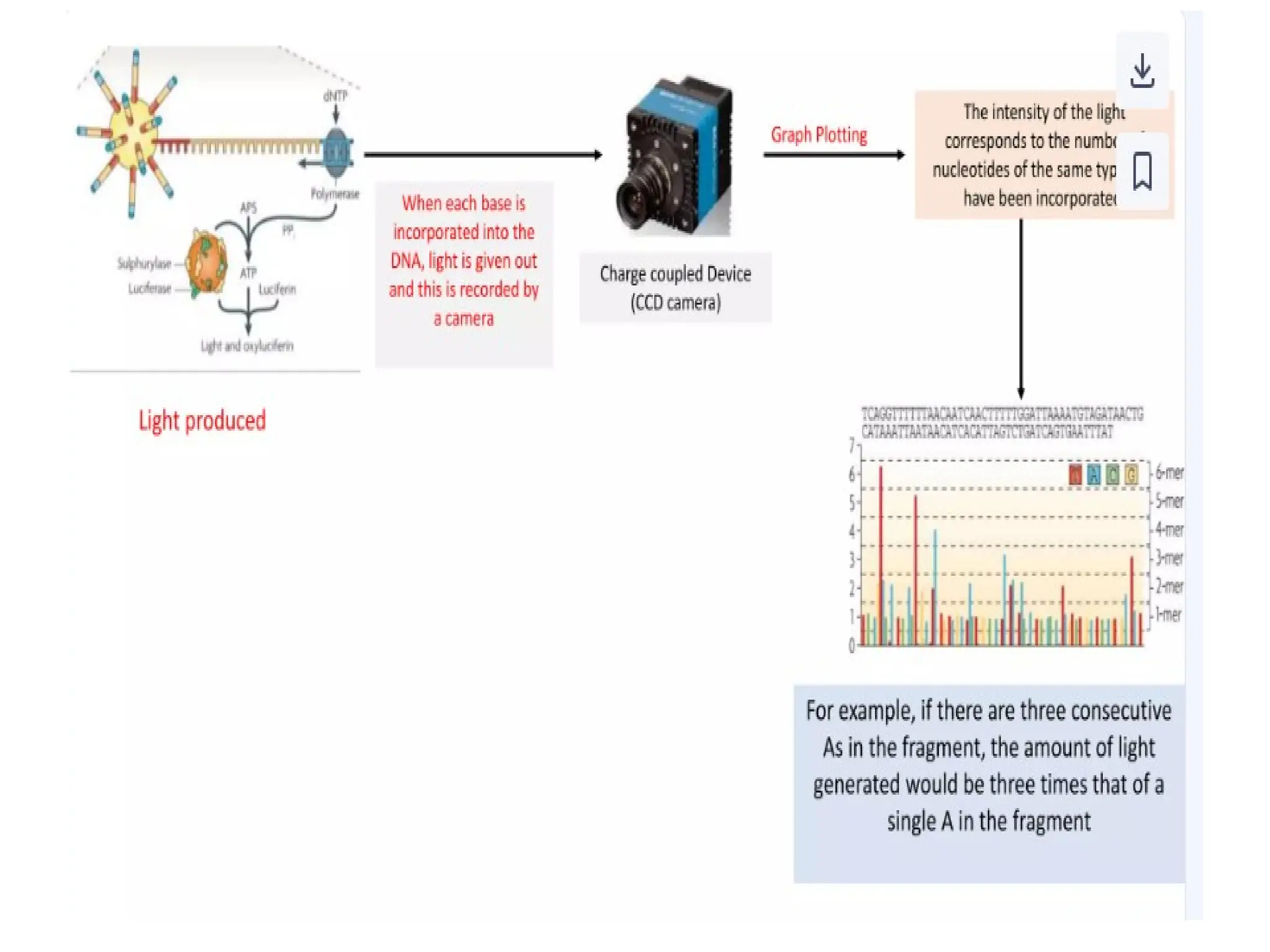
62.
62
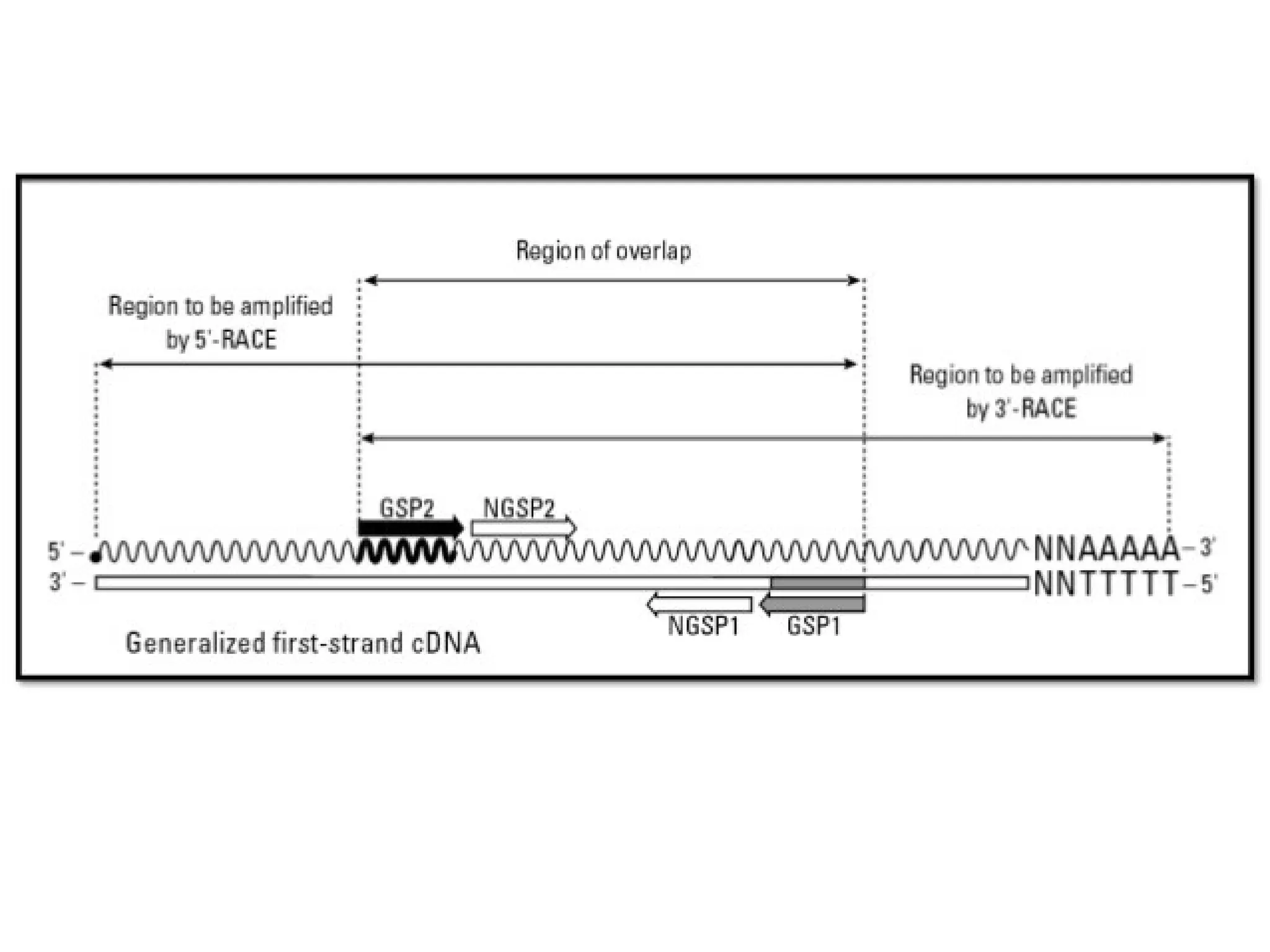
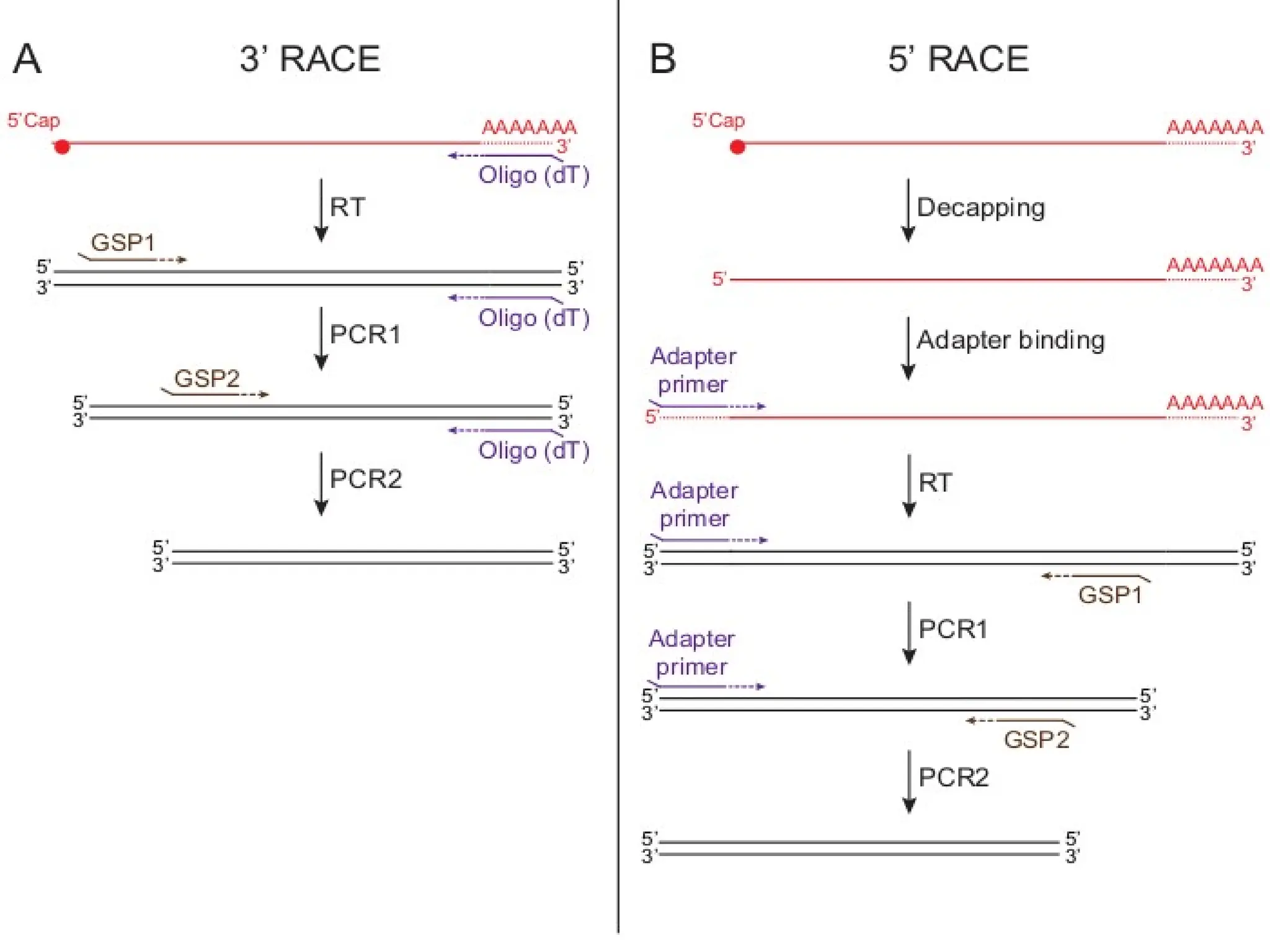
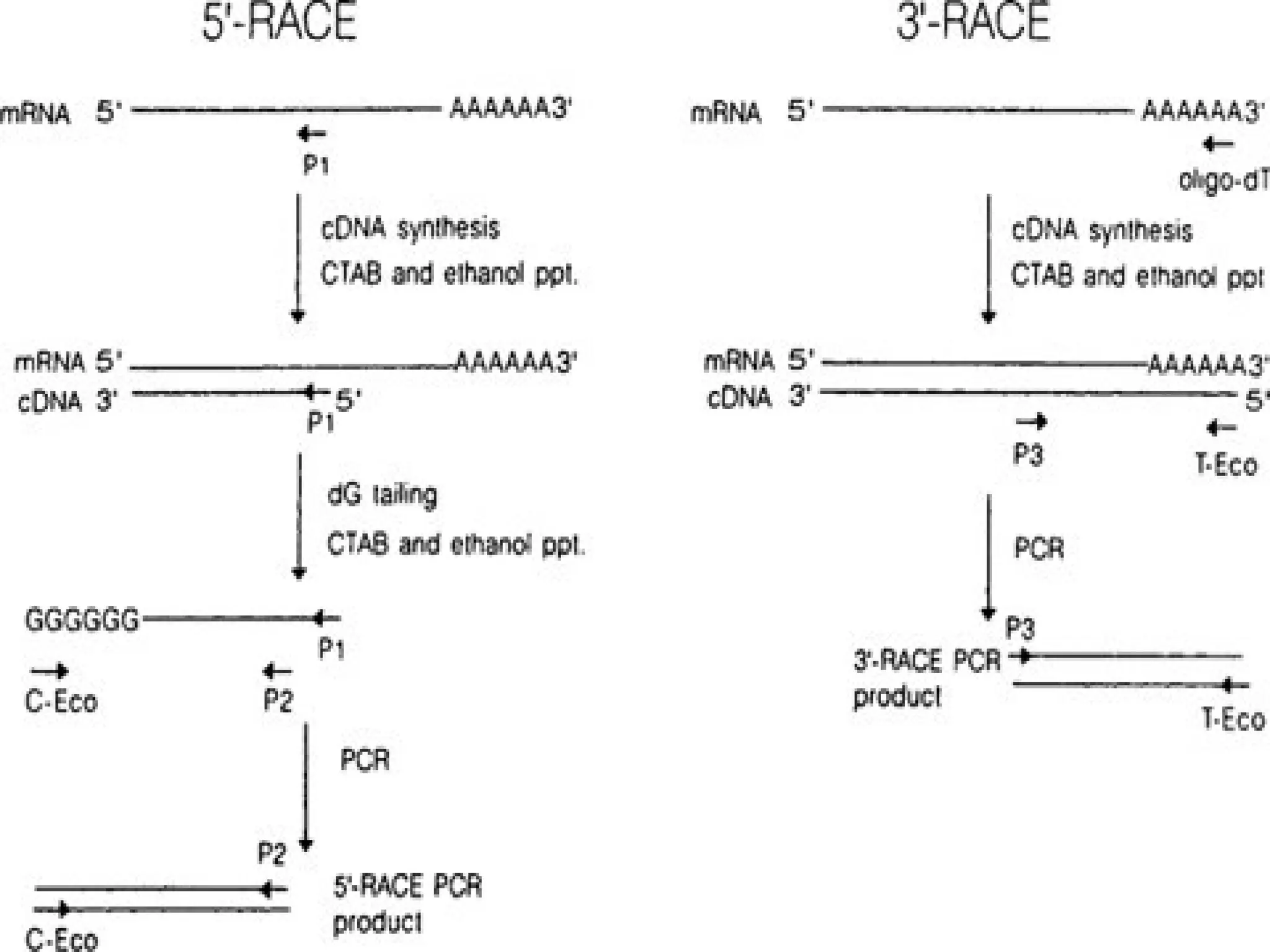
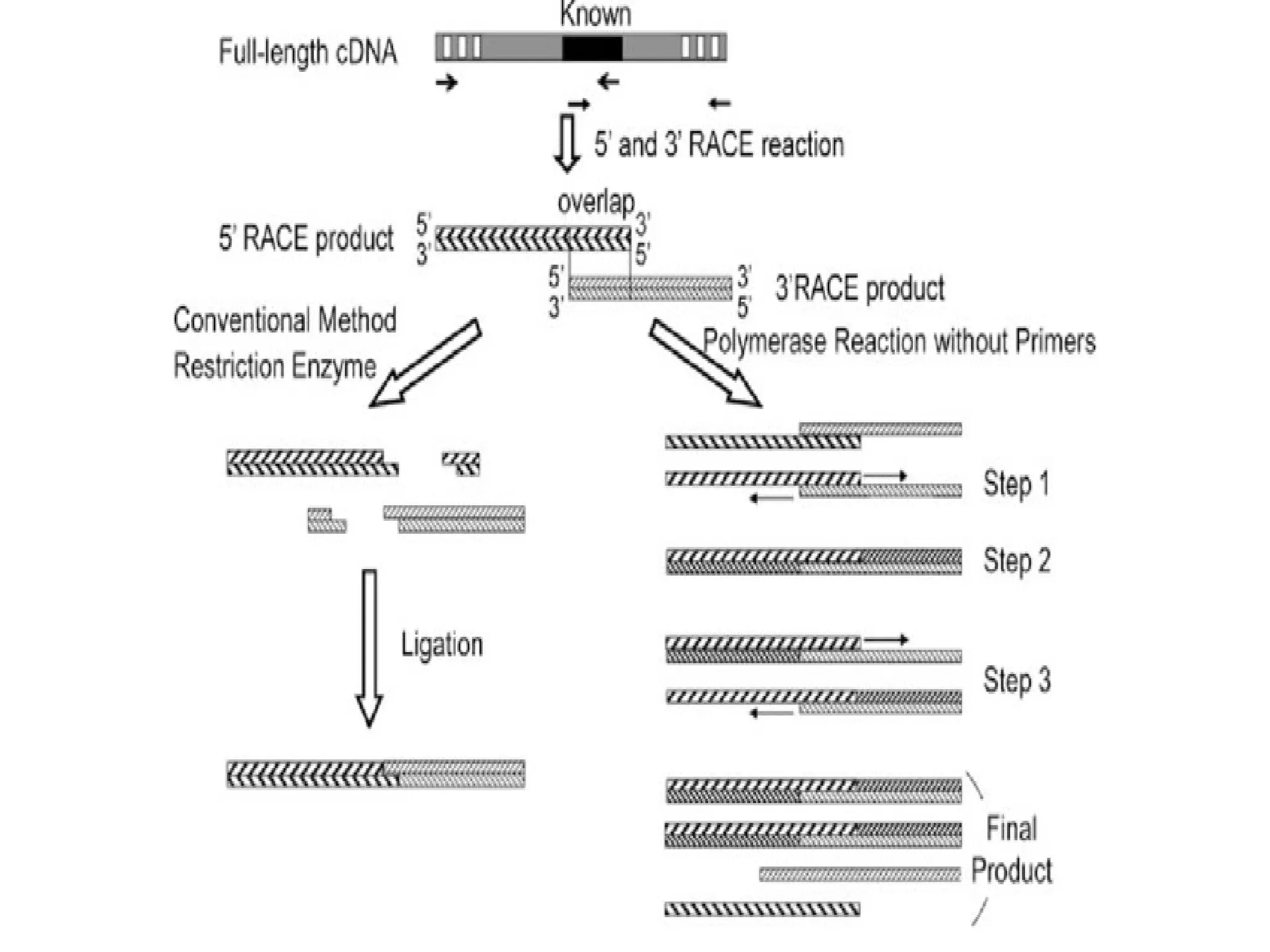
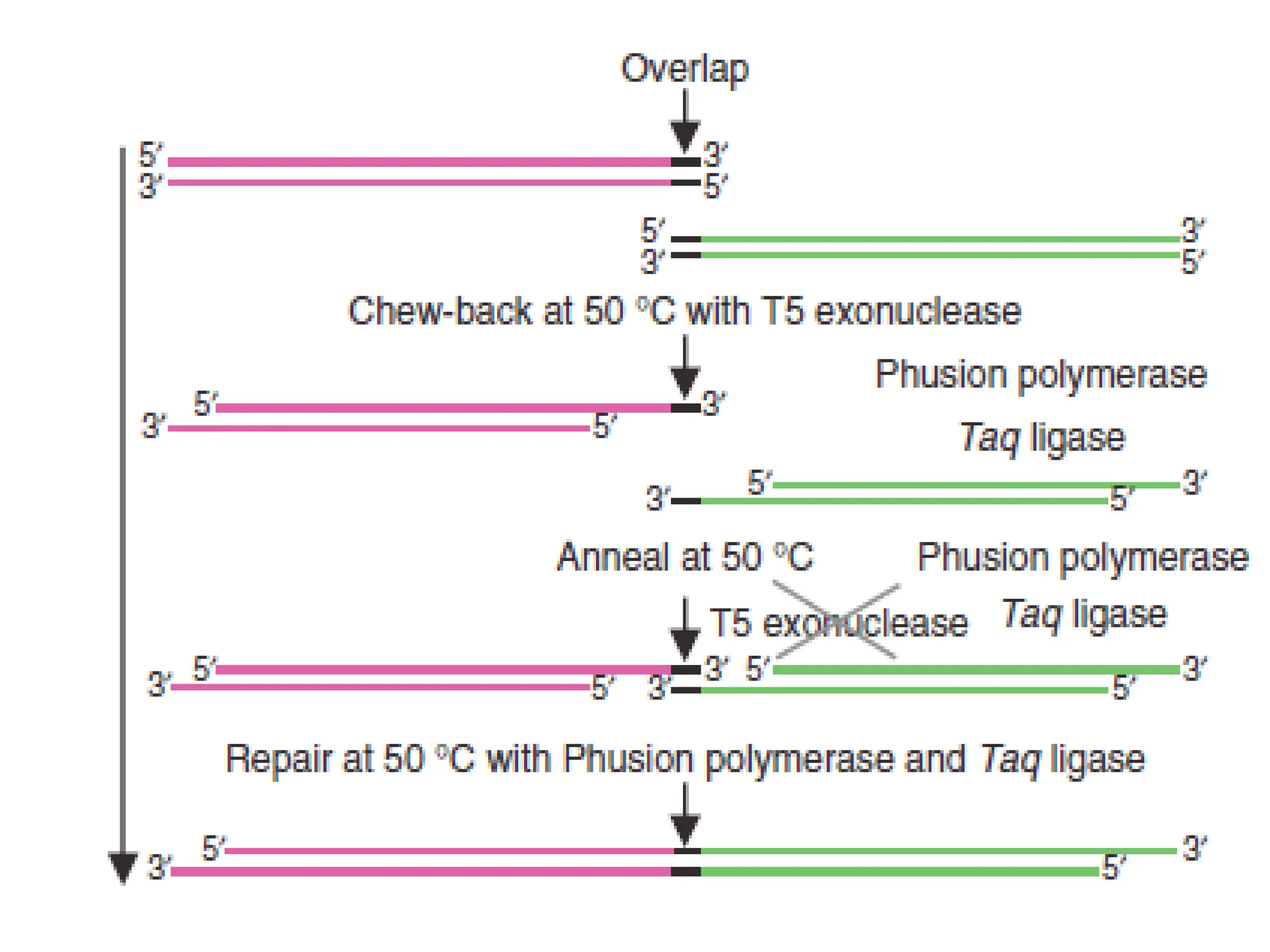
Procedure of 5’RACE
• Total or poly (A) RNA is reverse transcribed and first strand of cDNA is
synthesized by using a reverse gene-specific primer termed GSP 1 and
SuperScript™ II, a derivative of Mo-MLV RT with reduced RNase H
activity.
• After first cDNA strand synthesis, the original mRNA template is removed
by treatment with the RNase Mix (mixture of RNase H, which is specific
for RNA:DNA heteroduplex molecules and RNase Tl).
• cDNA is purified to get rid of unincorporated dNTPs, GSP 1, and proteins.
• A poly-A tail is then added to the 3'-end of the cDNA using TdT (terminal
deoxynucleotidyl transferase) and a dATP.
• A PCR reaction is then carried out using a second gene specific
primer (GSP2) that binds to the known sequence and a forward dT
primer that binds the homopolymeric tail added to the 3' ends of
the cDNAs to amplify a cDNA product from the 5' end of mRNA.
65
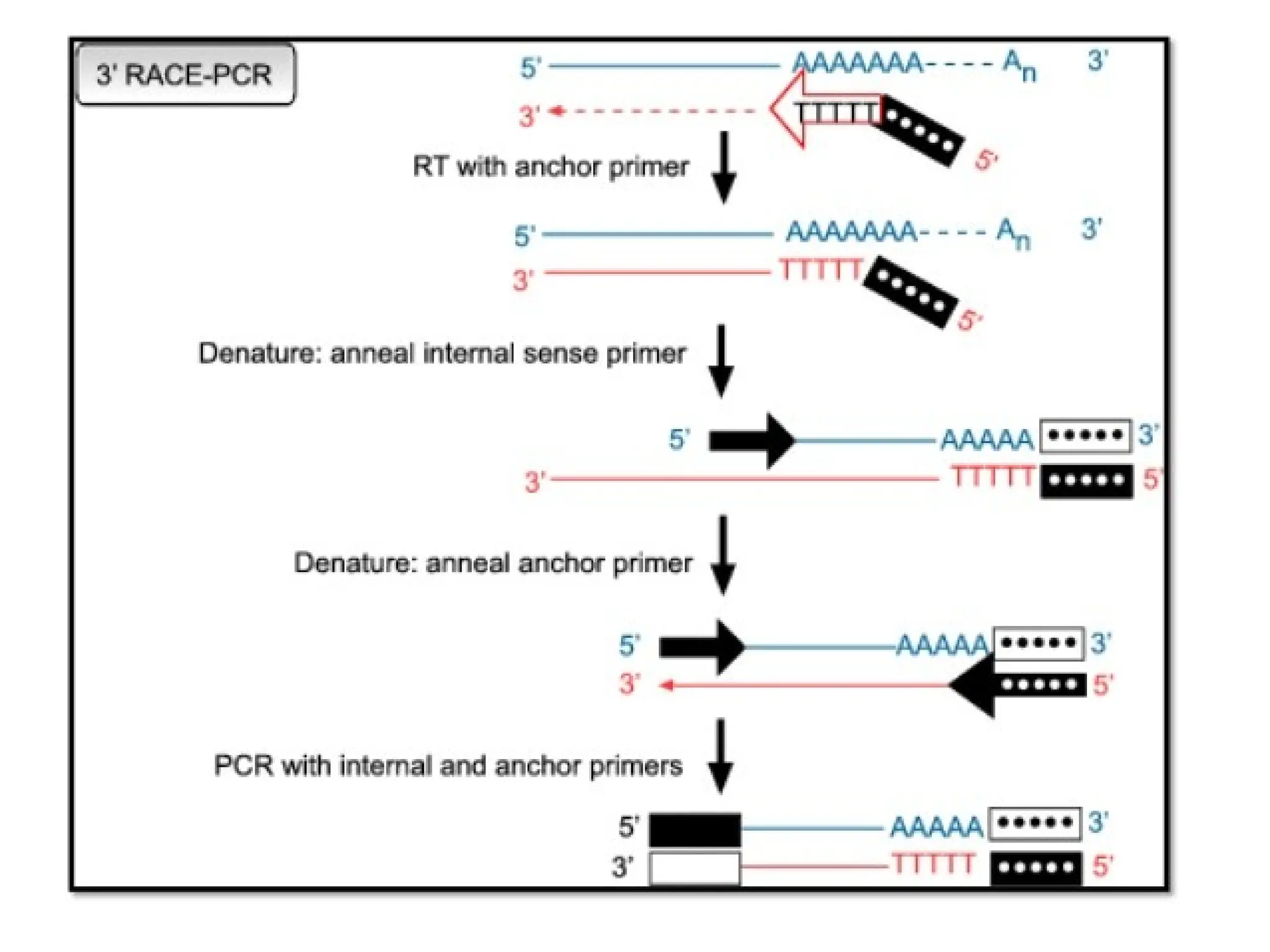
Procedure of 3'RACE
• First DNA strand is synthesized by reverse transcription of mRNA,
which is initiated at the poly (A) tail of mRNA using the adapter
primer (AP).
• After first cDNA strand synthesis, the original mRNA template is
degraded with Rnase H, which is specific for RNA:DNA
heteroduplex molecules.
• Then
amplification
is performed, without intermediate
phenol:chloroform extractions or ethanol precipitations, with the help
of two primers:
→ One is a user-designed GSP that anneals to a site located within
the cDNA molecule.
→The other is a universal amplification primer that targets the
mRNA of the cDNA complementary to the 3'end of the mRNA as
discussed in 5' RACE.
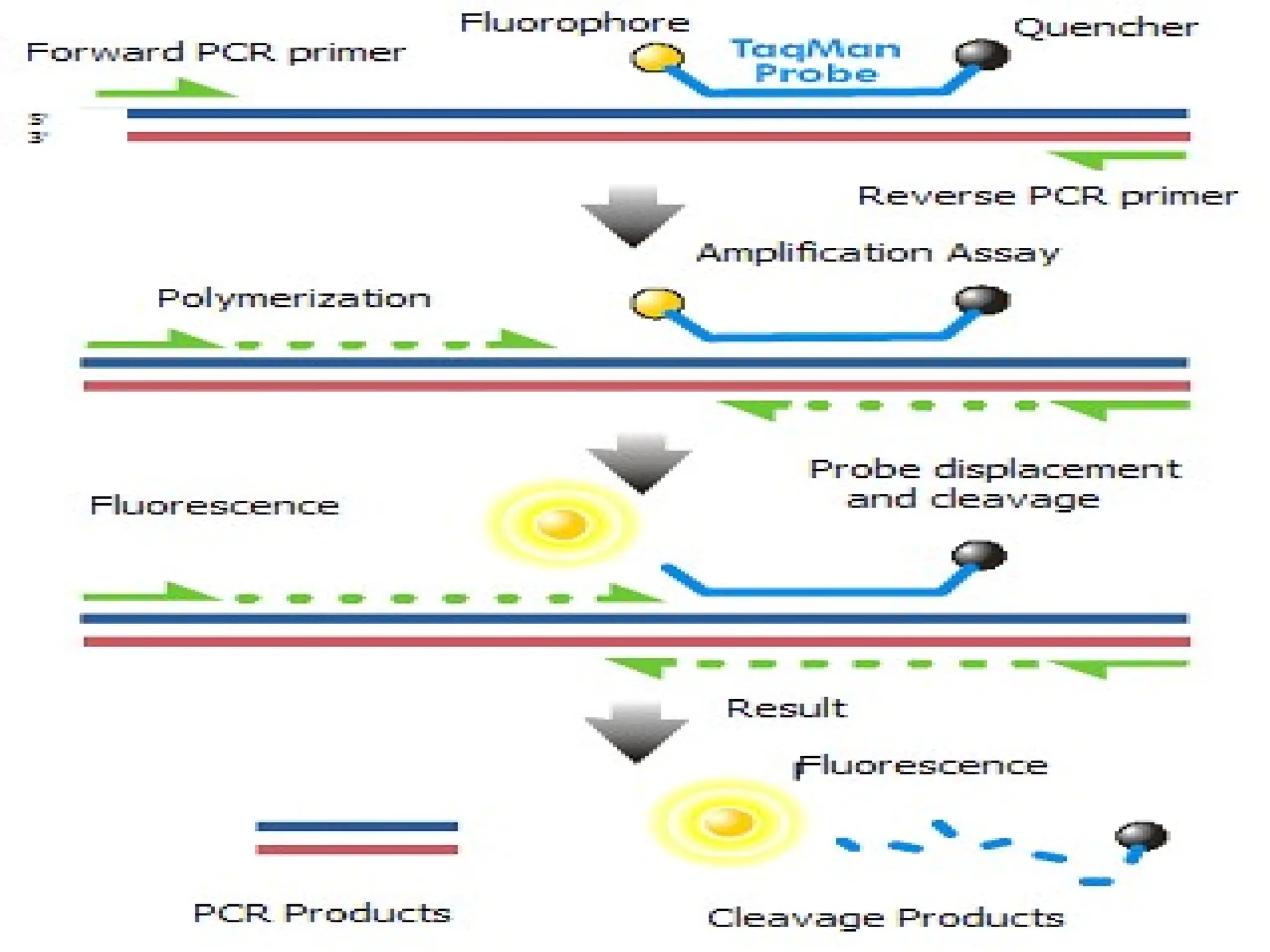
What is RealTime PCR?
amplicons to produce fluorescence during PCR.
The fluorescence, measured in Real Time, is
detected in a PCR cycler with an inbuilt filter
flurometer.
Real Time PCR is a
technique
in
which
fluoroprobes bind to specific target regions
of
87.
What are Fluorescentdyes?
When a population of fluorochrome molecules is excited by light of an
appropriate wavelength, fluorescent light is emitted. The light intensity
can be measured by flurometer or a pixel-by-pixel digital image of the
sample.
Excitation and Emission: Fluorodyes absorb light at one wavelength &
thereby boosts an electron to a higher energy shell.
•The excited electron falls back to the ground state and the flurophore re-
emits light but at longer wavelength.
•This shift makes it possible to separate excitation light from emission light
with the use of optical filters.
•The wavelength (nm) where photon energy is most efficiently captured is
defined as the Absorbancemax & the wavelength (nm) where light is most
efficiently released is defined as the Emissionmax.
90.
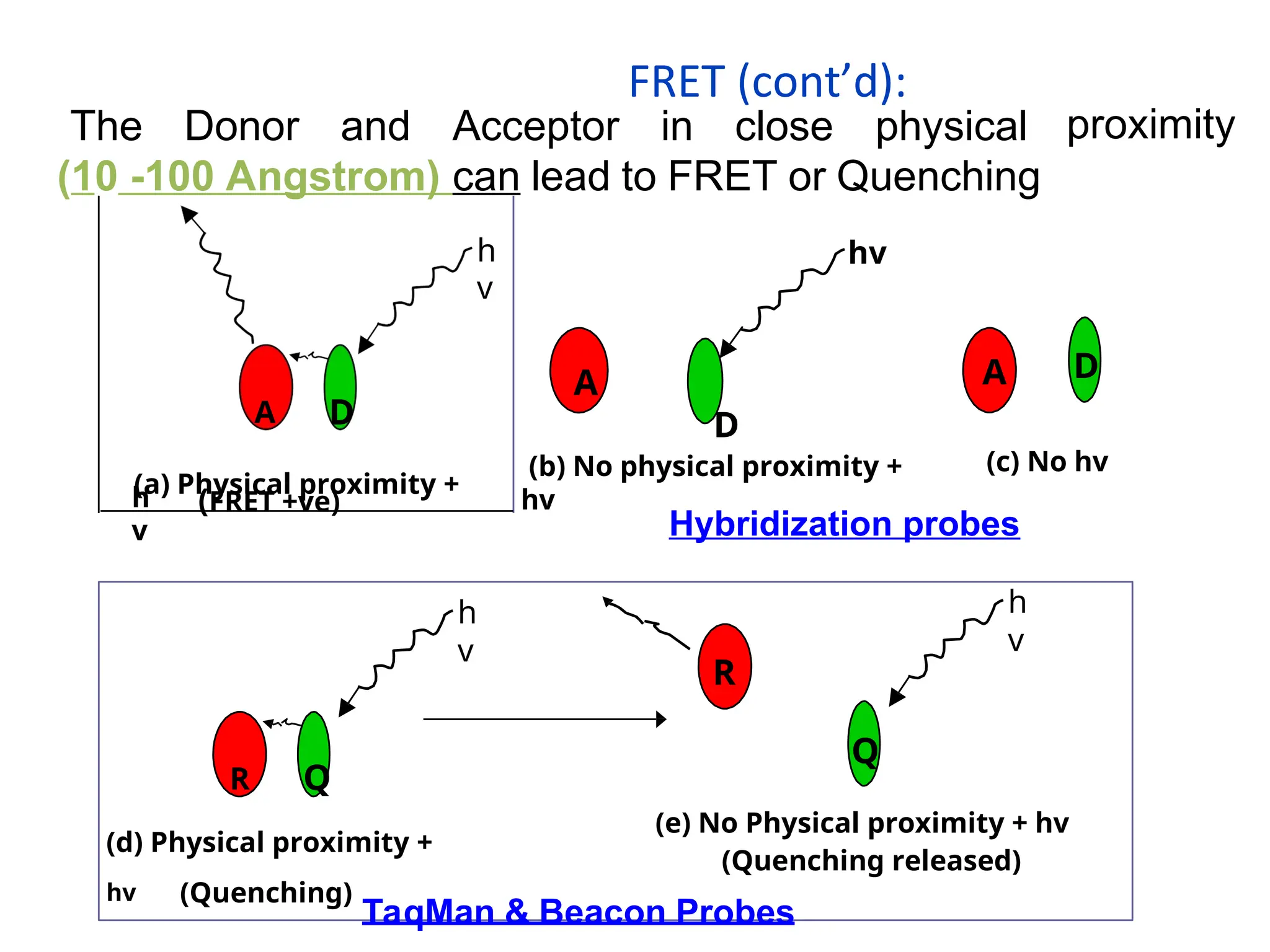
What is FluorescenceResonance Energy
Transfer (FRET)?
FRET is a distance dependent
interaction between the excited states
of 2 dye molecules in which excitation
is transferred from a donor molecule to
an acceptor molecule without emission of a
photon
The Donor andAcceptor in close physical
(10 -100 Angstrom) can lead to FRET or Quenching
proximity
hv
D
(b) No physical proximity +
hv
D
(c) No hv
R Q
(d) Physical proximity +
hv (Quenching)
h
v
(e) No Physical proximity + hv
(Quenching released)
R
h
v
Q
h
v
A D
(a) Physical proximity +
h
v
(FRET +ve)
Hybridization probes
TaqMan & Beacon Probes
A A
FRET (cont’d):
98.
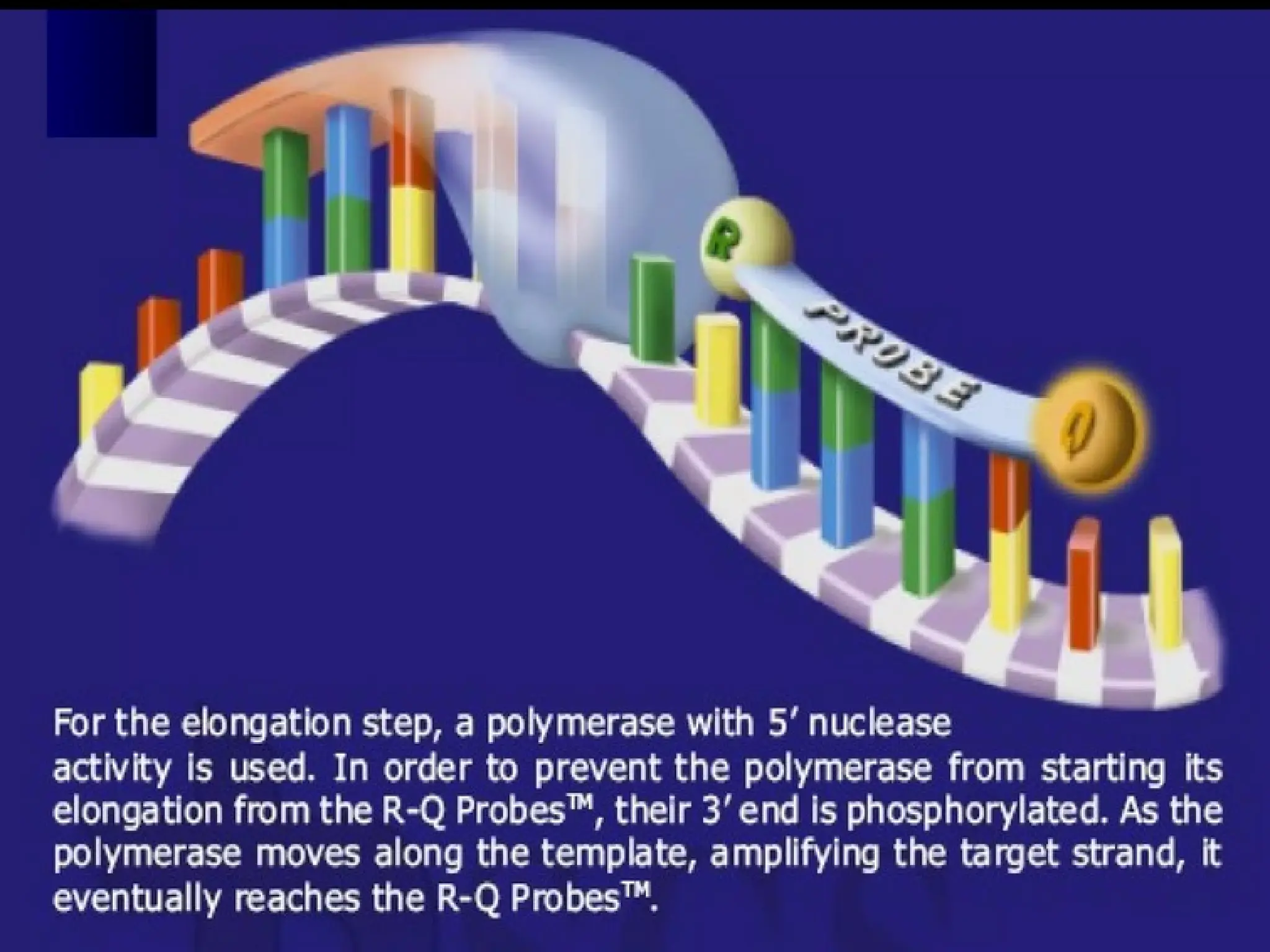
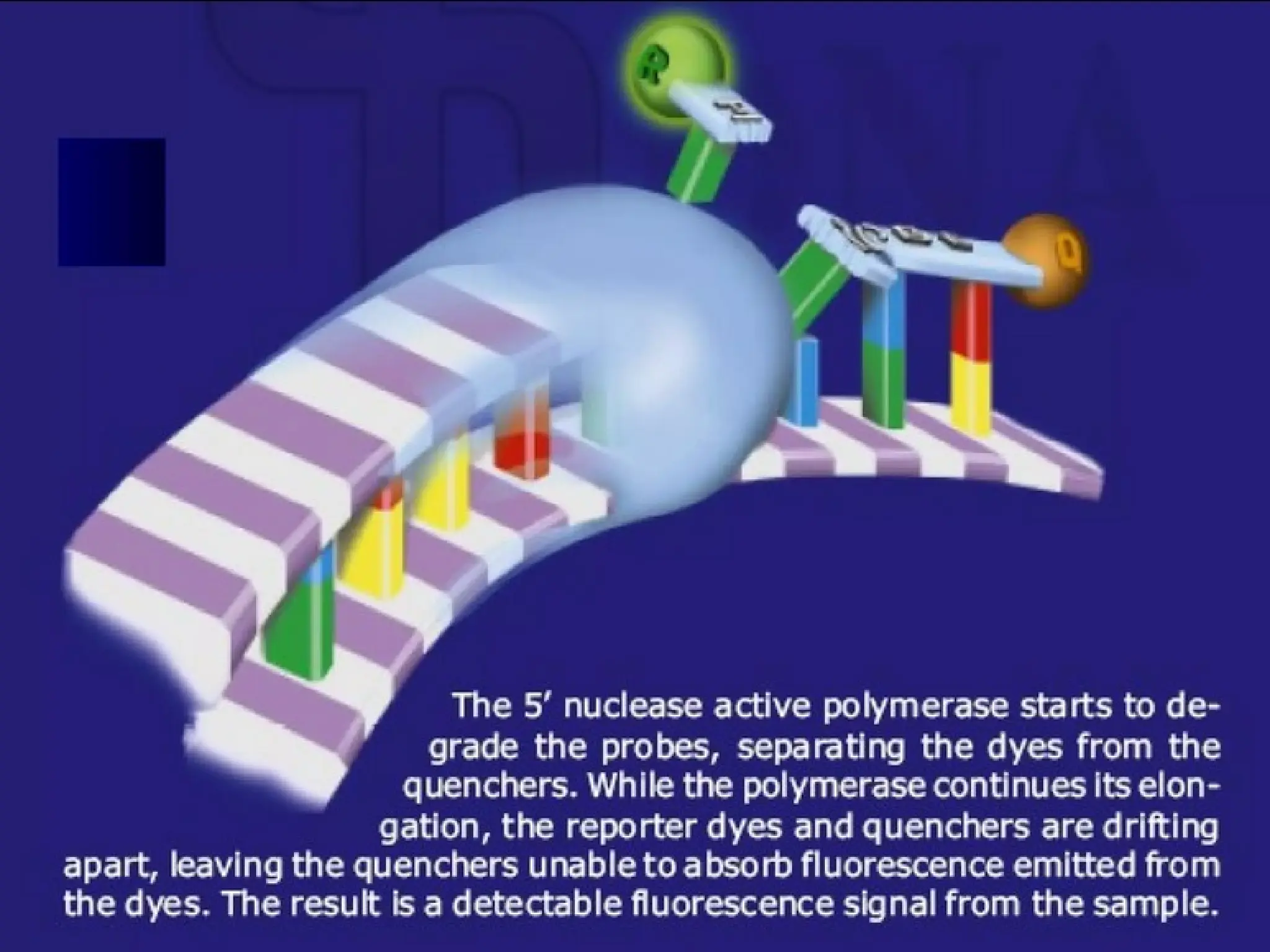
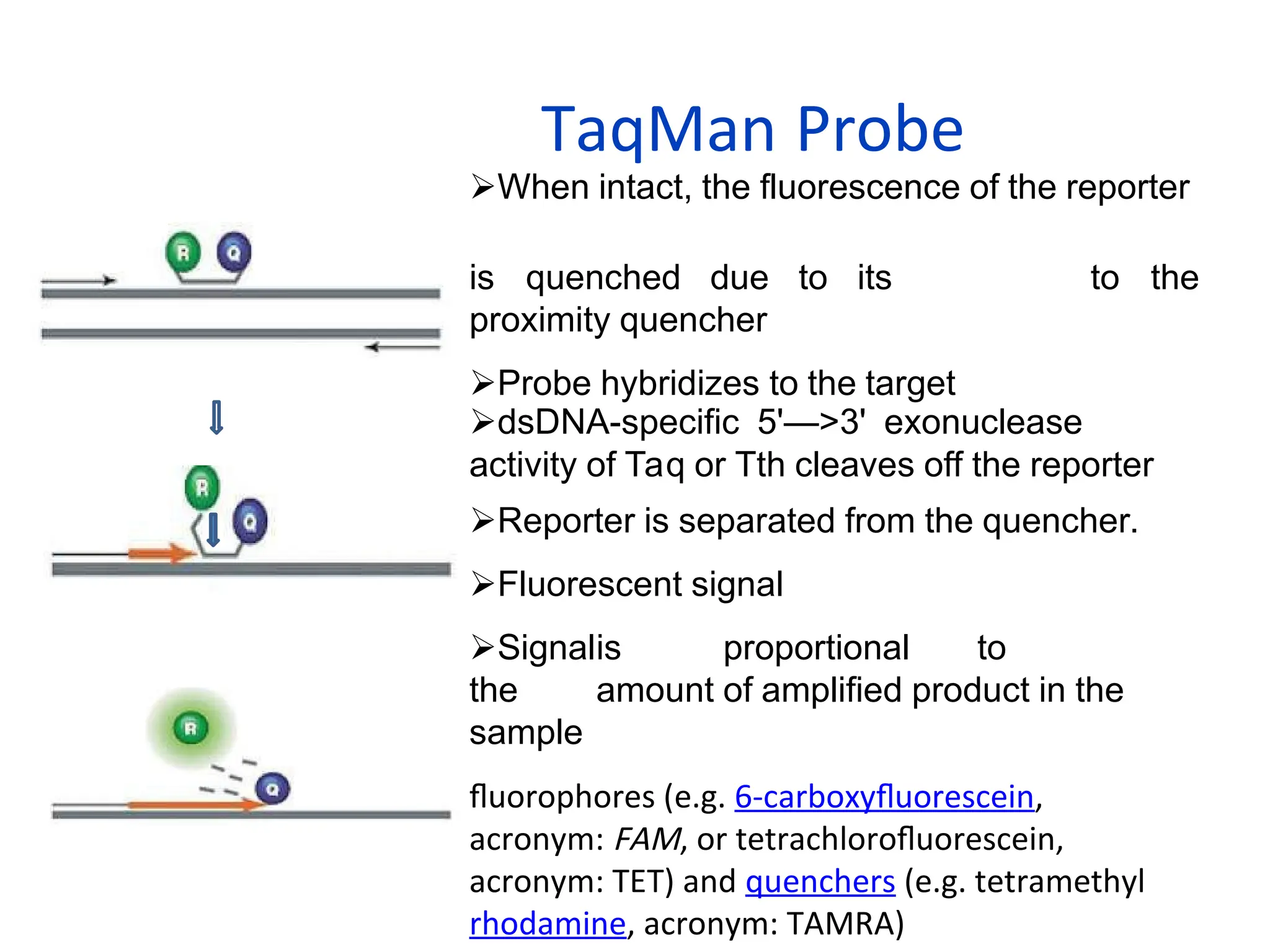
When intact, thefluorescence of the reporter
is quenched due to its
proximity quencher
Probe hybridizes to the target
to the
dsDNA-specific 5'—>3' exonuclease
activity of Taq or Tth cleaves off the reporter
Reporter is separated from the quencher.
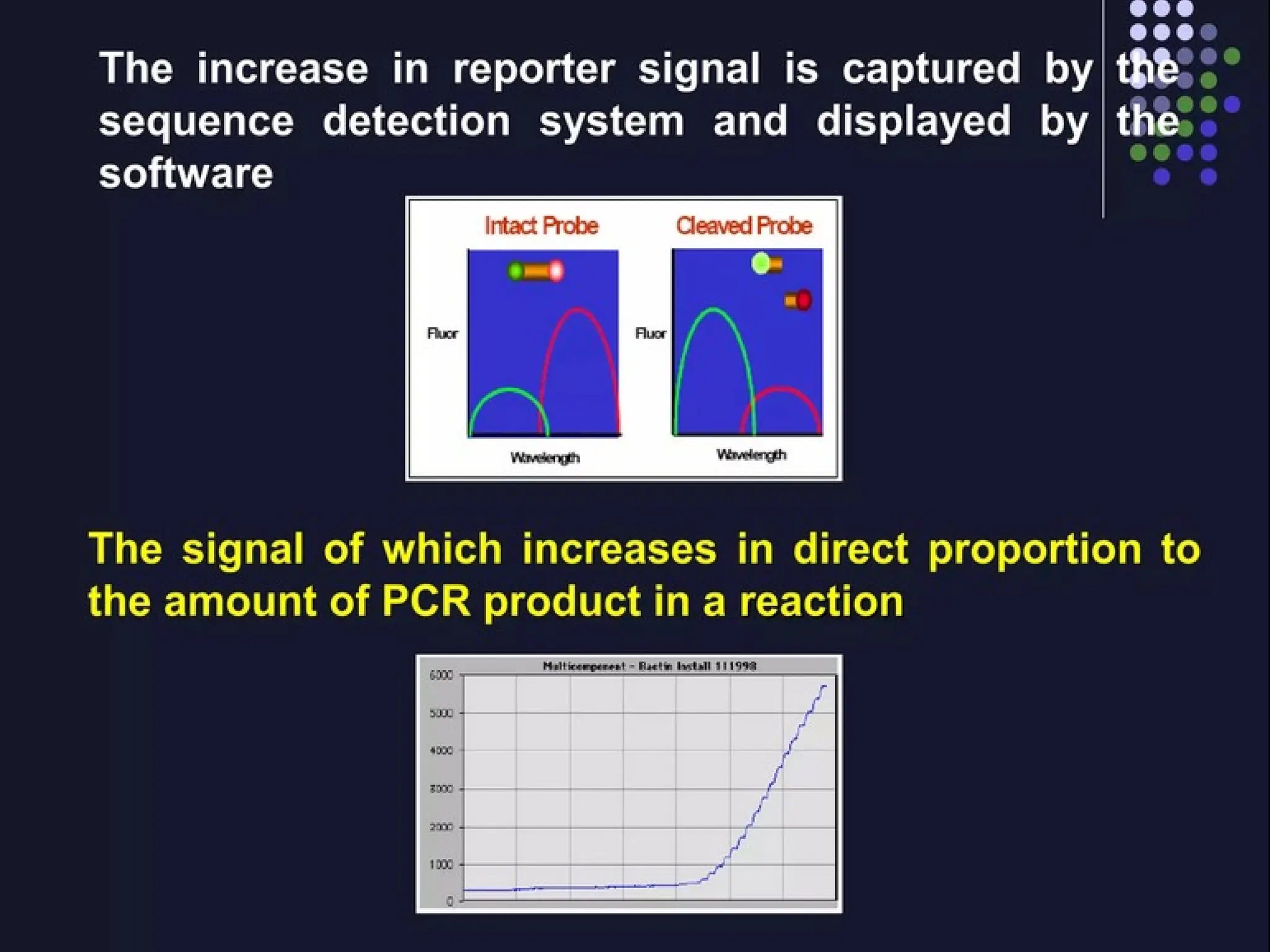
Fluorescent signal
Signalis proportional to
the amount of amplified product in the
sample
fluorophores (e.g. 6-carboxyfluorescein,
acronym: FAM, or tetrachlorofluorescein,
acronym: TET) and quenchers (e.g. tetramethyl
rhodamine, acronym: TAMRA)
TaqMan Probe
100.
Advantages
Highly fluorogenic
Easy PCRsetup
Sequence-specific detection, multiplexing
Disadvantages
Expensive
Probe design and positioning challenging
Similar conditions for primers and probes
Elevated background (Quenching capacity)
Probe degraded: no end-point analysis
TaqMan Probe
101.
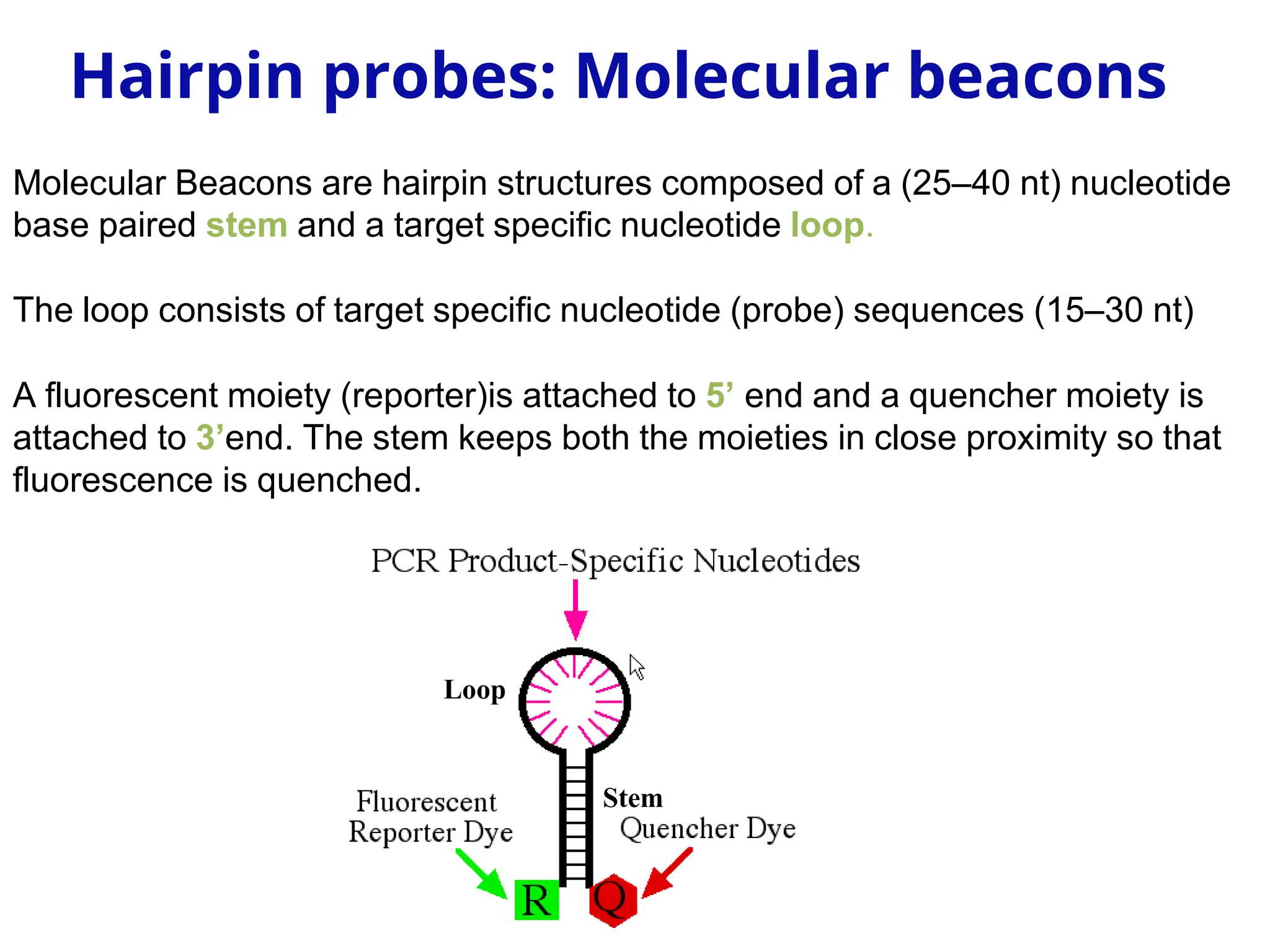
Loop
Stem
Molecular Beacons arehairpin structures composed of a (25–40 nt) nucleotide
base paired stem and a target specific nucleotide loop.
The loop consists of target specific nucleotide (probe) sequences (15–30 nt)
A fluorescent moiety (reporter)is attached to 5’ end and a quencher moiety is
attached to 3’end. The stem keeps both the moieties in close proximity so that
fluorescence is quenched.
Hairpin probes: Molecular beacons
102.
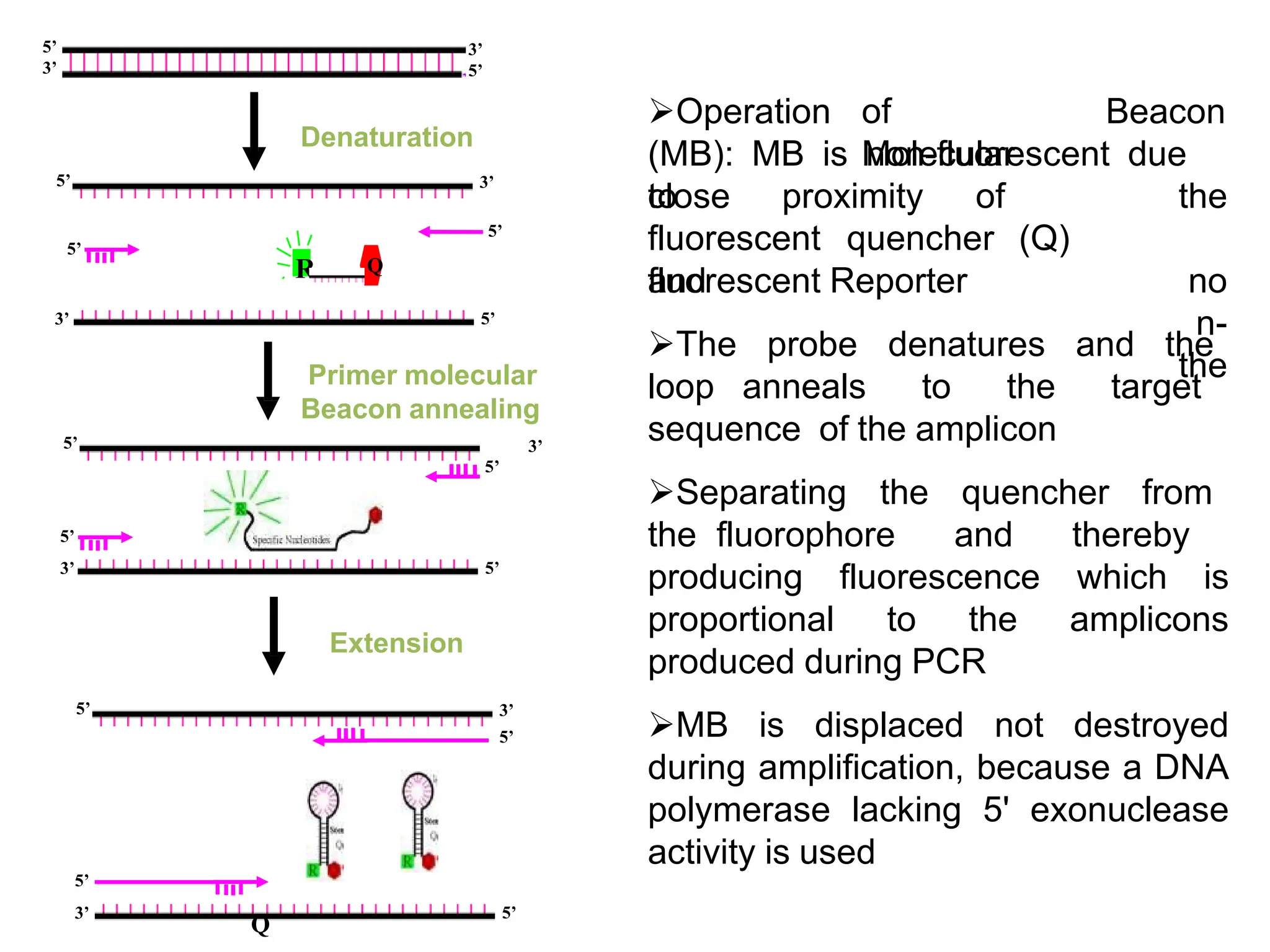
Denaturation
Primer molecular
Beacon annealing
3’
Extension
5’
5’
3’
Q
3’
5’
5’
5’
3’
3’
5’
5’
3’
3’
5’
5’
5’
5’
5’
3’
5’
Q
R
Operationof
Molecular
Beacon
(MB): MB is non-fluorescent due
to
close proximity of the
no
n-
the
fluorescent quencher (Q)
and
fluorescent Reporter
The probe denatures and the
loop anneals to the target
sequence of the amplicon
Separating the quencher from
the fluorophore and thereby
producing fluorescence which is
proportional to the amplicons
produced during PCR
MB is displaced not destroyed
during amplification, because a DNA
polymerase lacking 5' exonuclease
activity is used
107.
Molecular Beacons
Advantages
High specificity,low background
Post PCR analysis
PCR multiplex
Allelic discrimination (greater specificity than linear probes)
Disadvantages
Challenging design
Long probes – less yield
Intramolecular competitive binding
Low signal levels (proximity of reporter and quencher)
113.
• The Kunkelmethod for site-directed mutagenesis is a classic method
for introducing mutations (either single base pairs or larger
insertions, deletions, or substitutions) into a DNA sequence.
• There are three main steps to performing Kunkel mutagenesis.
• The dut gene encodes dUTPase which normally degrades dUTP.
An elevated concentration of dUTP accumulates in dut strains,
resulting in incorporation of U in place T at some positions during
DNA replication. The ung gene encodes uracil N-glycosylase which
normally removes U from DNA. Thus, in the double mutant U is
occasionally incorporated into DNA and this error is not repaired.
Because U has the same base pairing properties and the same coding
properties as T, incorporation of U into DNA in place of T is not
mutagenic.
115.
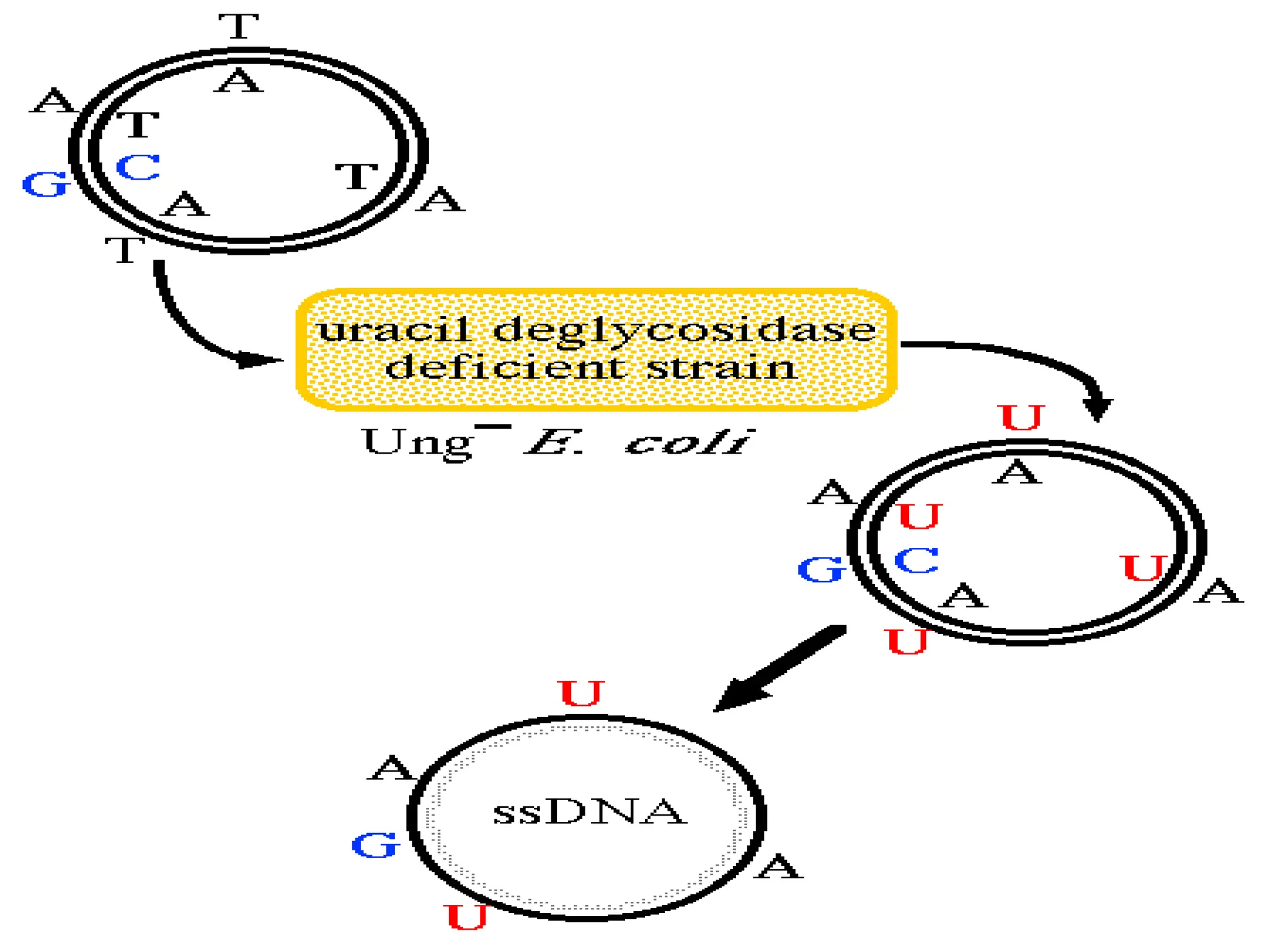
• In VivoProcess
1) Place the plasmid that contains your target
sequence to be mutated into an ung-
dut-
strain
of E. coli bacteria. dut-
(lacking dUTPase) bacteria
accumulate dUTP. ung-
(lacking uracil
deglycosidase) bacteria cannot remove dUTP that
gets incorporated into new DNA strands. The end
result is that your plasmid is converted to DNA that
lacks T's and contains U's instead:
117.
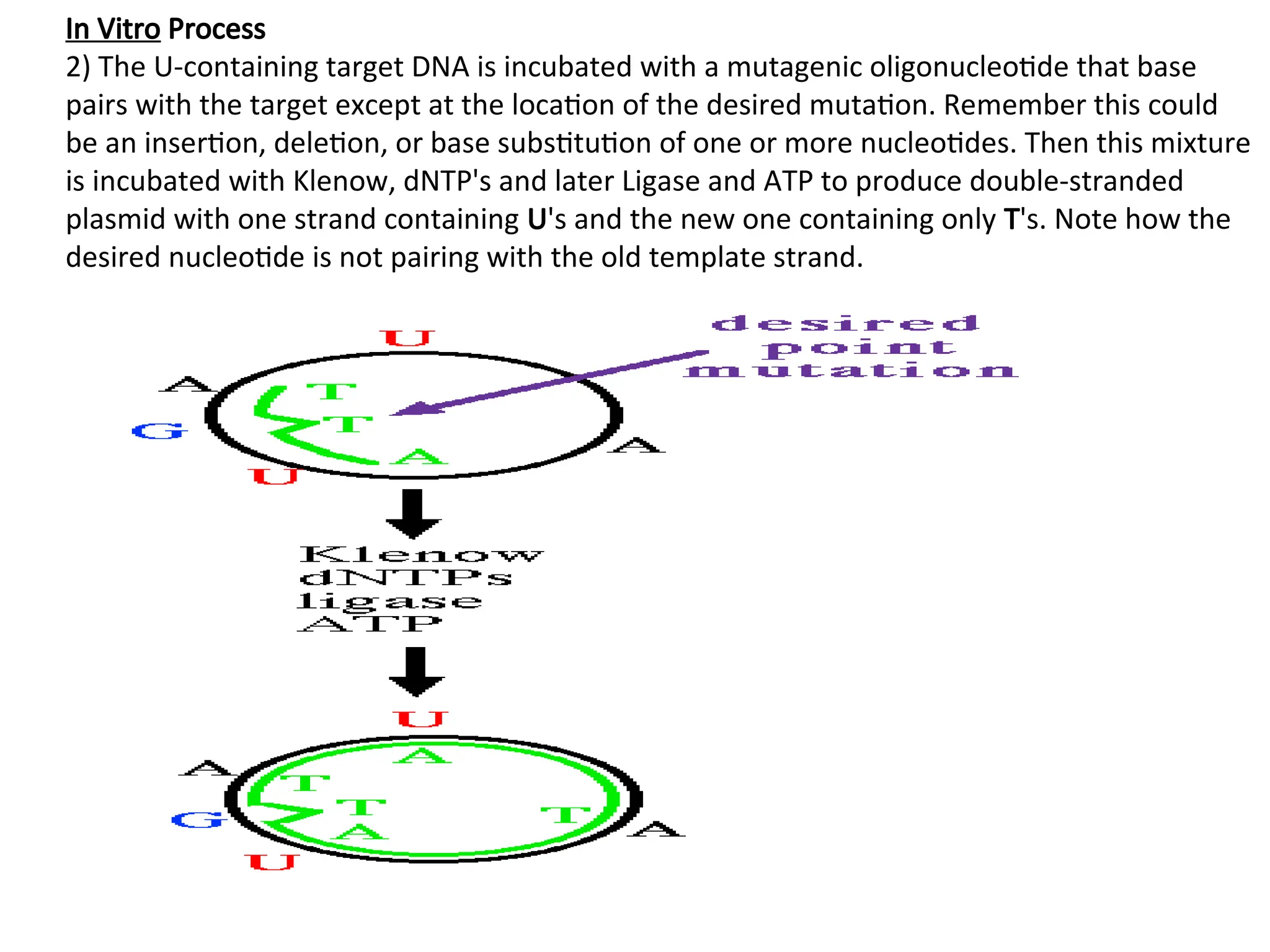
In Vitro Process
2)The U-containing target DNA is incubated with a mutagenic oligonucleotide that base
pairs with the target except at the location of the desired mutation. Remember this could
be an insertion, deletion, or base substitution of one or more nucleotides. Then this mixture
is incubated with Klenow, dNTP's and later Ligase and ATP to produce double-stranded
plasmid with one strand containing U's and the new one containing only T's. Note how the
desired nucleotide is not pairing with the old template strand.
118.
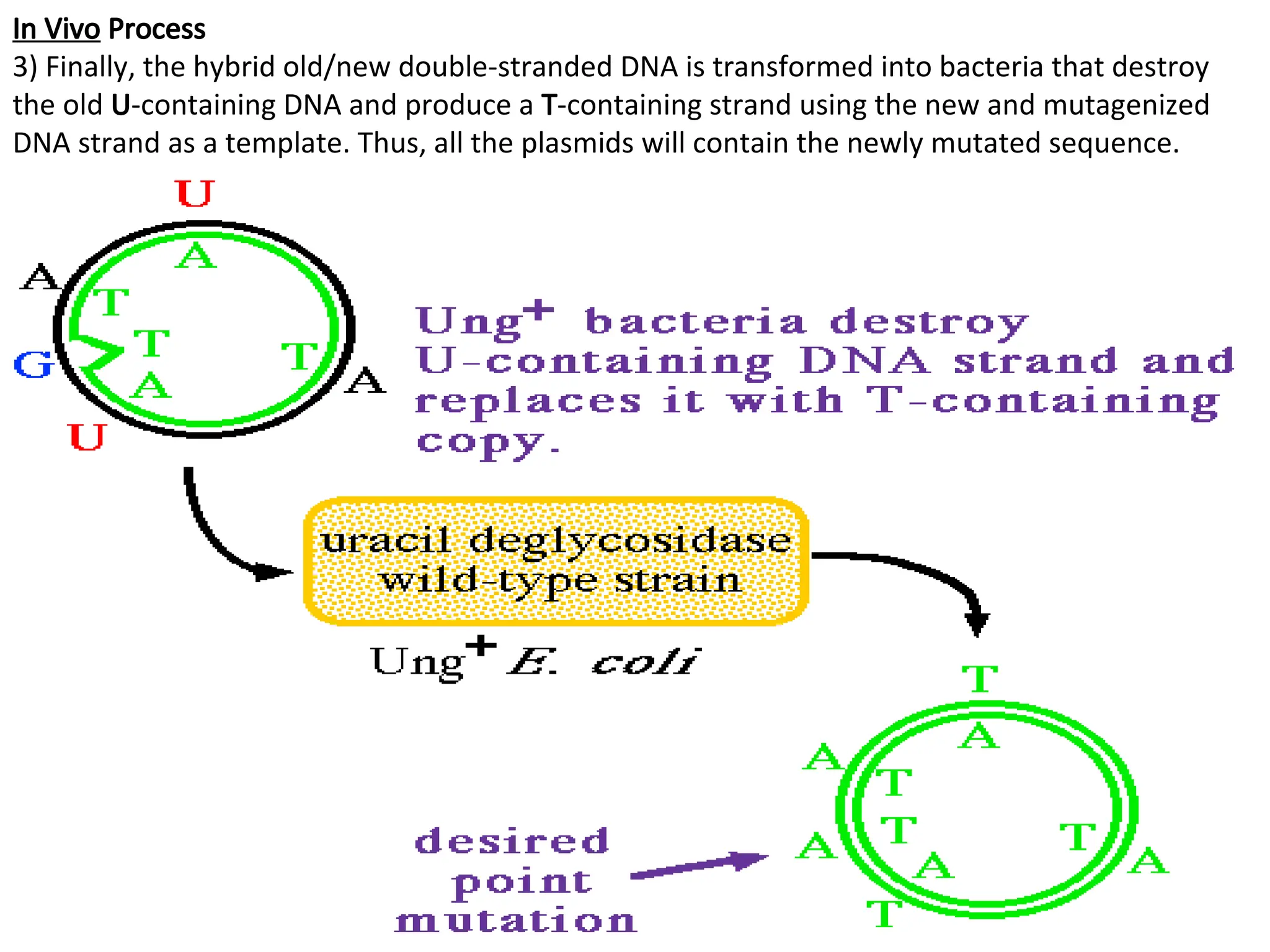
In Vivo Process
3)Finally, the hybrid old/new double-stranded DNA is transformed into bacteria that destroy
the old U-containing DNA and produce a T-containing strand using the new and mutagenized
DNA strand as a template. Thus, all the plasmids will contain the newly mutated sequence.