3 /38
3
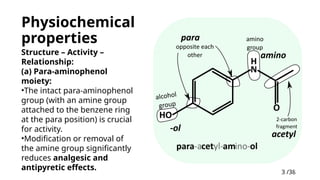
Physiochemical
properties
Structure –Activity –
Relationship:
(a) Para-aminophenol
moiety:
•The intact para-aminophenol
group (with an amine group
attached to the benzene ring
at the para position) is crucial
for activity.
•Modification or removal of
the amine group significantly
reduces analgesic and
antipyretic effects.
4.
4 /38
4
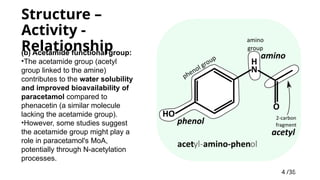
Structure –
Activity-
Relationship
(b) Acetamide functional group:
•The acetamide group (acetyl
group linked to the amine)
contributes to the water solubility
and improved bioavailability of
paracetamol compared to
phenacetin (a similar molecule
lacking the acetamide group).
•However, some studies suggest
the acetamide group might play a
role in paracetamol's MoA,
potentially through N-acetylation
processes.
5.
5 /38
5
Structure –
Activity-
Relationship
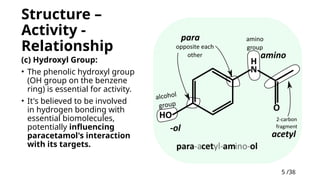
(c) Hydroxyl Group:
• The phenolic hydroxyl group
(OH group on the benzene
ring) is essential for activity.
• It's believed to be involved
in hydrogen bonding with
essential biomolecules,
potentially influencing
paracetamol's interaction
with its targets.
6.
6 /38
6
Other physiochemicalproperties
• pH: approximately 6
• Storage: Store in a cool, dry place away from direct sunlight and
heat. High temperatures can degrade the medication.
• Stability: Dry, pure paracetamol exhibits good stability at
temperatures up to 45°C. However, contamination with traces of
para-aminophenol or exposure to humid conditions can lead to
hydrolysis and degradation, resulting in discoloration.
• Isomerism and its properties:
1. Para-aminophenol (amino group in 4th
position) Paracetamol derived
from this
2. Ortho-aminophenol no medical use
7.
7 /38
7
Pharmacokinetics
Distribution:
•Plasma halflife = 1.5-2.5 hours
•Onset of action around 40 mins
(oral), 8 mins (IV)
•Protein binding – negligible (15-
20% in case of overdose)
•Large volume of distribution (50
L)
•Widely distributed throughout the
body tissues, with high penetration
into the central nervous system
(CNS)
•Log P (Octanol-Water Partition
Coefficient): Around 0.5. This
value suggests moderate
lipophilicity, allowing for passive
diffusion across cell membranes,
Absorption:
•Route: Oral or IV or IM or rectal
•Rapid from small intestine,
usually high (but variable)
bioavailability
•Dose-dependent Bioavailability
63% (500 mg) to 89% (1000 mg)
•pKa: Approximately 9.4. This
value indicates minimal pH-
dependent solubility within the
physiological range (pH 7.3-7.4).
8.
8 /38
8
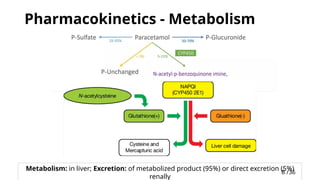
Pharmacokinetics -Metabolism
Metabolism: in liver; Excretion: of metabolized product (95%) or direct excretion (5%)
renally
10 /38
10
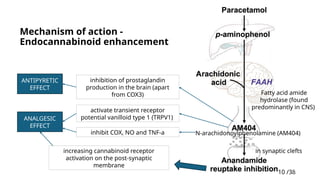
Mechanism ofaction -
Endocannabinoid enhancement
increasing cannabinoid receptor
activation on the post-synaptic
membrane
Fatty acid amide
hydrolase (found
predominantly in CNS)
N-arachidonoylphenolamine (AM404)
in synaptic clefts
activate transient receptor
potential vanilloid type 1 (TRPV1)
inhibition of prostaglandin
production in the brain (apart
from COX3)
inhibit COX, NO and TNF-a
ANALGESIC
EFFECT
ANTIPYRETIC
EFFECT
11.
11 /38
11
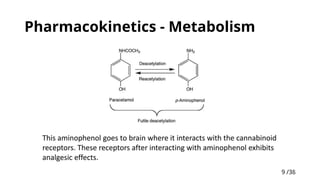
Mechanism ofaction:
POX and COX inhibition
(This enzyme has two
active sites (POX –
Peroxidase, and COX)
In brain, a splice variant of COX-1
(termed as COX-3) was present,
which was inhibited by paracetamol
Paracetamol acts as reducing
substrate for POX (decreases its
regeneration)
(this is peroxide-level dependent
reaction: In periphery due to
inflammation peroxides are
usually present; compared to
periphery, peroxide levels are low in
the brain since they cannot cross
BBB)
12.
12 /38
12
Mechanism ofaction - Serotoninergic pathway activation
PARACETAMOL
activation of descending
serotonergic pathways
(originating in the brainstem
nuclei, hypothalamus, and cortex,
and interact with pain afferents in
the dorsal horn)
ANALGESIC EFFECT
13.
13 /38
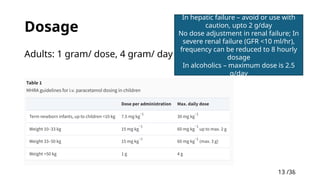
13
Dosage
Adults: 1gram/ dose, 4 gram/ day
In hepatic failure – avoid or use with
caution, upto 2 g/day
No dose adjustment in renal failure; In
severe renal failure (GFR <10 ml/hr),
frequency can be reduced to 8 hourly
dosage
In alcoholics – maximum dose is 2.5
g/day
14.
14 /38
14
In pregnancy
•No dose adjustment is required
• Oral/ rectal – category B; IV – category C
15.
15 /38
15
Uses
• Fever
•Headache
• Musculoskeletal pain like Osteoarthritis
• Dental pain
• Back ache
• Dysmenorrhoea
16.
16 /38
16
Uses inAnaesthesia
1. Multimodal Analgesia – with opioids and NSAID’s
2. Premedication with paracetamol (oral or IV) before
anesthesia induction can:
• Minimize propofol injection pain, a common complaint during induction.
• Improve surgical tolerance by reducing intraoperative awareness.
3. Post operative shivering - IV paracetamol has central
thermoregulatory effects, helping to prevent and treat
postoperative shivering.
4. Post operative Nausea and Vomiting (PONV) (inconsistent
evidence)
17.
17 /38
17
Novel uses– IV Regional anaesthesia
augmentation
• During IV regional anaesthesia,
adding paracetamol to the
injected lidocaine improve the
overall quality of the block.
• In view of the overall consensus
that paracetamol’s actions are
centrally mediated, an analgesic
benefit conferred from its addition
to a peripherally sequestered pool
of drug is a surprising finding.
• Onset of motor block was
sooner
• Tourniquet pain was
reduced
• Recovery of motor and
sensory block was
delayed
• resulting in lower
intraoperative pain
scores and
• Decreased total systemic
analgesic requirements.
19 /38
19
Adverse events
•Paracetamol can be associated with non-specific
gastrointestinal symptoms, such as nausea and vomiting,
dyspepsia, abdominal pain, and bloating. (long term users
irritation of mucosa)
20.
20 /38
20
Paracetamol toxicity
•Toxic dose: >150 mg/kg (or) >10 gram in adult
• Fatality is common with >250 mg/kg
• In certain individuals, doses in less than usual toxic dose can
cause toxicity, secondary to deficiencies in glutathione
because of:
a) inadequate nutrition
b) CYP2E1 enzyme induction by chronic alcohol excess, or concomitant
use of other drugs.
• Paracetamol toxicity can cause acute liver failure. This can be
associated with acute renal failure
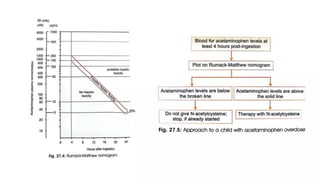
• Acute poisoning has 4 stages
24 /38
24
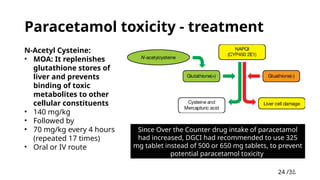
Paracetamol toxicity- treatment
N-Acetyl Cysteine:
• MOA: It replenishes
glutathione stores of
liver and prevents
binding of toxic
metabolites to other
cellular constituents
• 140 mg/kg
• Followed by
• 70 mg/kg every 4 hours
(repeated 17 times)
• Oral or IV route
Since Over the Counter drug intake of paracetamol
had increased, DGCI had recommended to use 325
mg tablet instead of 500 or 650 mg tablets, to prevent
potential paracetamol toxicity
26 /38
26
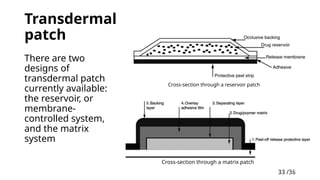
Transdermal patch
•The transdermal route for drug delivery avoids first pass
metabolism and large variations in plasma drug
concentrations.
• Drugs suitable for transdermal administration have a low
molecular weight and high lipid solubility.
• There are two types of patches available: reservoir and
matrix systems.
27.
27 /38
27
Skin andTDDS
1.Skin structure and permeability:
The stratum corneum, the
outermost layer of the epidermis, is
the primary barrier to drug
absorption. This highly lipophilic
membrane, with low water content
(20%), impedes penetration of
foreign molecules.
2.Drug absorption pathway:
Drugs must traverse the lipophilic
and hydrophilic domains of the
stratum corneum to reach the
dermis, where a dense capillary
network facilitates rapid systemic
absorption.
28.
28 /38
28
Skin andTDDS
3.Alternative pathways:
Hydrophilic drugs can utilize
pathways like hair follicles,
sweat glands, and blood
vessels for dermal delivery.
However, these routes have
minimal contribution to steady-
state drug flux in the systemic
circulation.
4.Factors influencing
absorption:
Individual variations in stratum
corneum thickness, skin
hydration, underlying skin
conditions, ethnicity, and body
temperature can affect the rate
of percutaneous drug
absorption.
29.
29 /38
29
Pharmacokinetics ofTDDS
Drug release and absorption:
• The TDDS acts as a drug depot, storing the drug in a reservoir or
within its matrix.
• Upon application, a concentration gradient drives drug movement
from the patch towards the dermis.
• A secondary reservoir is established within the stratum corneum.
• The drug is then absorbed into the local capillaries and enters
systemic circulation.
• This process leads to a delay (variable by drug) in achieving the
desired minimum effective concentration (MEC) in plasma, with
undetectable levels initially.
30.
30 /38
30
Pharmacokinetics ofTDDS
Steady-state and duration of action:
• Reaching steady-state plasma concentration can take time, potentially
requiring multiple patch applications (2-3). Once achieved, this
concentration is maintained for the duration of patch use.
• TDDS offers the benefit of continuous drug delivery, improving
patient compliance.
Drug depletion after removal:
• Following patch removal, drug concentrations decline gradually.
• The rate of decline depends on the drug's context-sensitive half-life
and the presence of a residual skin reservoir.
31.
31 /38
31
Effect ofdrug characteristics
All of the drugs
currently available in
patch formulation
share three features :
1. molecular mass,
<500 Da;
2. high lipophilicity;
and
3. low required daily
dose (<2 mg).
33 /38
33
Transdermal
patch
Cross-section througha reservoir patch
Cross-section through a matrix patch
There are two
designs of
transdermal patch
currently available:
the reservoir, or
membrane-
controlled system,
and the matrix
system
34.
34 /38
34
Reservoir patchvs Matrix patch
Reservoir patch Matrix patch
holds the drug in a gel or solution incorporates the drug into an adhesive
polymer matrix.
Delivery dose is determined by a rate-
controlling membrane between the
drug reservoir and the skin slow rate
of drug release (less interpatient
variability)
Delivered dose depends on the amount
of drug held, and area of the patch
applied.
formulation of the drug/polymer matrix
slow rate of drug release
Reservoir patches give tighter control of
delivery rates but can have an initial
burst of drug release.
The active ingredient is distributed evenly
throughout the patch. One-half of a patch
will have half the original surface area and
deliver half the original dose per hour.
Accidental overdose if the membrane is
damaged which causes risk of sudden
Less risk of accidental overdose and offers
less potential for abuse
35.
35 /38
35
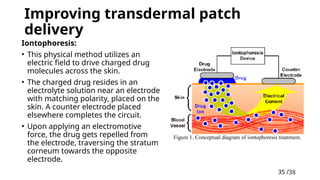
Improving transdermalpatch
delivery
Iontophoresis:
• This physical method utilizes an
electric field to drive charged drug
molecules across the skin.
• The charged drug resides in an
electrolyte solution near an electrode
with matching polarity, placed on the
skin. A counter electrode placed
elsewhere completes the circuit.
• Upon applying an electromotive
force, the drug gets repelled from
the electrode, traversing the stratum
corneum towards the opposite
electrode.
36.
36 /38
36
Improving transdermalpatch
delivery
Iontophoresis:
• This movement also generates
convective flow within the solvent,
dragging neutral drug molecules
along (electro-
osmosis).Additionally, electric
current might transiently enhance
skin permeability.
• This technique allows for delivering
bolus doses of drugs and has been
utilized in the development of
fentanyl patient-controlled
analgesia (PCA) patches.
Chemical enhancement
Examples include adding
solvents like ethanol or
propylene glycol to increase
drug solubility and facilitate
permeation via stratum
corneum
37.
37 /38
37
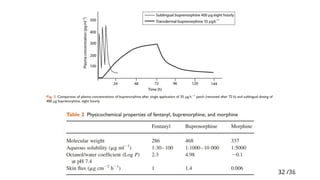
Buprenorphine TDDS(matrix)
• Buprenorphine is a partial agonist at μ-receptors; it is 60 times more
potent than morphine.
• A ceiling effect is reached at doses of >16 mg/ day. This does not
happen in clinical practice as the patches are designed to deliver 35,
52.5, or 70 μg/hr and the maximum dose is 3.36 mg/day (two 70 μg/hr
patches).
• Effective plasma concentrations are reached within 12 –24 h.
• Metabolism is by the CYP 3A4 system but offset after patch removal is
slower due to the high affinity of buprenorphine for opioid receptors.
• A recent development is the release of a 7 days buprenorphine patch.
This buprenorphine patch is a matrix system, available in three sizes,
delivering 5, 10, or 20 μg/hr over 7 days, and used for the treatment of
moderate to severe post-operative pain.
#6 This near-neutral pH makes it suitable for various pharmaceutical formulations and minimizes potential interactions with other drugs based on pH.
#7 IV paracetamol: Offers faster onset (within 5 minutes) and predictable plasma concentrations compared to oral and rectal routes, crucial in the perioperative period.
Oral Tablet, Syrup, Powder, Effervescent granules, Capsule
Rectal suppositories
Intravenous
Intramuscular
#10 . This would explain the experiences of relaxation, tranquility, and euphoria reported by many paracetamol users, apparently independent of analgesia.
#11 The exact MoA of paracetamol remains somewhat elusive, with both central and peripheral effects proposed.
#12 Serotonin receptors are present throughout the central nervous system, involved in a number of functions, including consciousness, mood, memory, and nausea and vomiting, the latter of which are mediated via the 5-HT3-receptor subtype. It has become widely accepted that the plays a key role in the action of paracetamol, and it has been demonstrated that the antinociceptive effects of paracetamol can be partially inhibited by co-administration of 5-HT3-receptor antagonists, interestingly using anti-emetic drugs which are indeed frequently given together with paracetamol in the perioperative period.
#16 Postoperative shivering is a common complication that can increase oxygen demand and discomfort.
#27 Epidermis thickness varies across the body (0.06-0.8 mm), impacting permeability.
#39 In general, paracetamol is thought to have only minor effects on renal function, of no clinical relevance in the vast majority of patients.
that said, acute tubular necrosis has been observed as an isolated finding in rare cases.
#40 In adult patients, the hypotension was associated with increased skin blood flow, consistent with its antipyretic action; these effects were not demonstrated in healthy afebrile volunteers, or in elective surgical patients when given paracetamol perioperatively.
Conversely, regular use of oral paracetamol has been linked with a raised blood pressure (studies: heart rate and blood pressure may show statistically, though perhaps not clinically, significant rises)
The limited evidence on the subject would suggest that adults and neonates in a critical care setting, who are either febrile or have pre-existing low blood pressure, may have increased susceptibility to a period of hypotension after either enteral or i.v. paracetamol. Whilst often only modest and brief, a proportion of these hypotensive episodes did require supportive intervention, although no long-term sequelae were reported.
Whilst much of these data come from retrospective observational studies, results from two small randomized, placebo-controlled crossover trials conducted in patients with known coronary artery disease or treated hypertension suggest that after as little as 2 weeks of paracetamol at submaximal doses of 1 g three times a day,
#41 In adult patients, the hypotension was associated with increased skin blood flow, consistent with its antipyretic action; these effects were not demonstrated in healthy afebrile volunteers, or in elective surgical patients when given paracetamol perioperatively.
Conversely, regular use of oral paracetamol has been linked with a raised blood pressure (studies: heart rate and blood pressure may show statistically, though perhaps not clinically, significant rises)
The limited evidence on the subject would suggest that adults and neonates in a critical care setting, who are either febrile or have pre-existing low blood pressure, may have increased susceptibility to a period of hypotension after either enteral or i.v. paracetamol. Whilst often only modest and brief, a proportion of these hypotensive episodes did require supportive intervention, although no long-term sequelae were reported.
Whilst much of these data come from retrospective observational studies, results from two small randomized, placebo-controlled crossover trials conducted in patients with known coronary artery disease or treated hypertension suggest that after as little as 2 weeks of paracetamol at submaximal doses of 1 g three times a day,
#43 In adult patients, the hypotension was associated with increased skin blood flow, consistent with its antipyretic action; these effects were not demonstrated in healthy afebrile volunteers, or in elective surgical patients when given paracetamol perioperatively.
Conversely, regular use of oral paracetamol has been linked with a raised blood pressure (studies: heart rate and blood pressure may show statistically, though perhaps not clinically, significant rises)
The limited evidence on the subject would suggest that adults and neonates in a critical care setting, who are either febrile or have pre-existing low blood pressure, may have increased susceptibility to a period of hypotension after either enteral or i.v. paracetamol. Whilst often only modest and brief, a proportion of these hypotensive episodes did require supportive intervention, although no long-term sequelae were reported.
Whilst much of these data come from retrospective observational studies, results from two small randomized, placebo-controlled crossover trials conducted in patients with known coronary artery disease or treated hypertension suggest that after as little as 2 weeks of paracetamol at submaximal doses of 1 g three times a day,