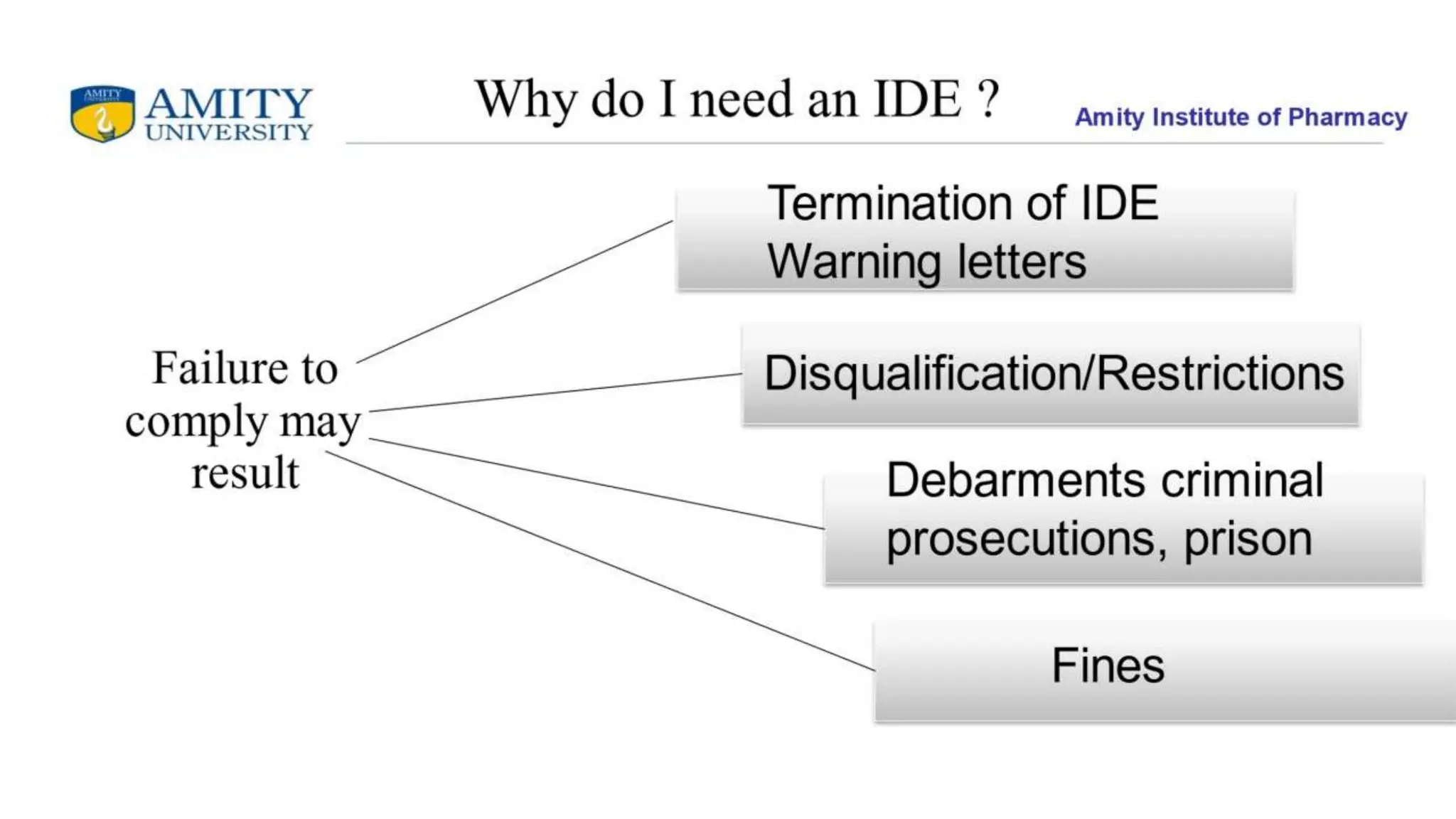
The document discusses the Investigational Device Exemption (IDE), which allows for clinical investigation of devices under FDA regulation. It also covers In Vitro Diagnostics (IVD) and their classification into risk-based regulatory classes, along with Quality System Requirements (QSR) that ensure product safety and effectiveness. Additionally, it outlines labeling requirements as specified in various parts of the Code of Federal Regulations.

















![European_Union.ppt.Nikhil[1].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/europeanunion-220803170320-4be1aa31-thumbnail.jpg?width=640&height=640&fit=bounds)































































