Download to read offline
















![In 3% to 7% of cases,[citation needed] Angelman syndrome occurs when
a person has two copies of the paternal chromosome 15 instead of one
copy from each parent. This
phenomenon is called paternal Uniparental disomy (UPD). People with
paternal UPD for chromosome 15 have two copies of the UBE3A gene, but
they are both inherited from
the father and are therefore inactive in the brain.
About 10% of Angelman syndrome cases are caused by a mutation in
the UBE3A gene, and
another 3% result from a defect in the DNA region that controls the
activation of
the UBE3A gene and other genes on the maternal copy of chromosome
15. In a small percentage of cases, Angelman syndrome may be caused
by a chromosomal
rearrangement called a translocation or by a mutation in a gene other than
UBE3A.
These genetic changes can abnormally inactivate the UBE3A gene.](https://image.slidesharecdn.com/humanchromosome15-180916072920/85/Human-chromosome-15-24-320.jpg)



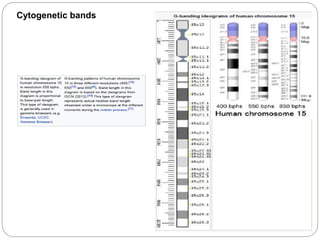
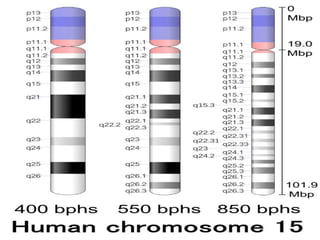
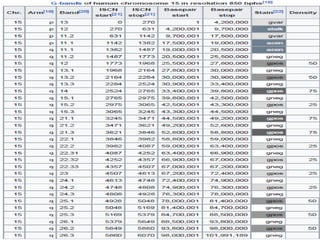
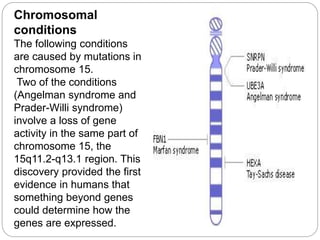
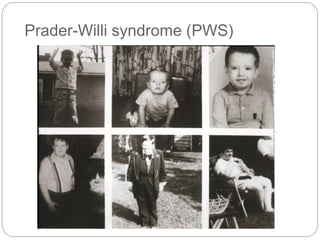
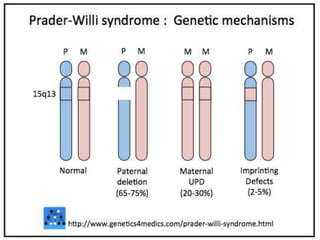
This document provides information about human chromosome 15. It discusses that chromosome 15 is an acrocentric chromosome that represents about 3% of human genetic material. It contains over 900 genes, including some linked to diseases. The sequence of the euchromatic region has been fully mapped. Conditions like Prader-Willi syndrome and Angelman syndrome are caused by deletions or defects involving imprinted genes on chromosome 15.






















































































