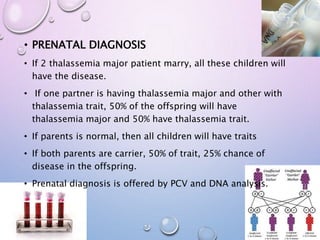
Downloaded 148 times






















This document summarizes haemoglobinopathies, specifically sickle cell disease and thalassemia. It describes the pathogenesis, diagnosis and management of sickle cell disease, including complications during pregnancy. It also describes different types of alpha and beta thalassemia based on number of affected globin genes, from asymptomatic trait to fatal forms incompatible with life. Prenatal diagnosis options are discussed for couples at risk of passing on severe haemoglobinopathies.


































































![Anemia in Pregnancy [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/anemiainpregnancyautosaved-220726145128-395be97a-thumbnail.jpg?width=640&height=640&fit=bounds)






























