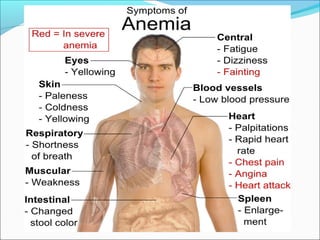
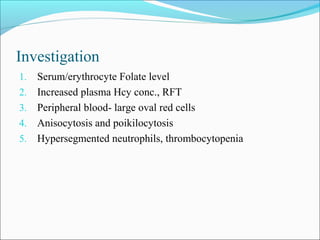
The document discusses vitamin B12 deficiency anemia. It defines anemia and describes the stages and pathophysiology of vitamin B12 deficiency. Key points include:
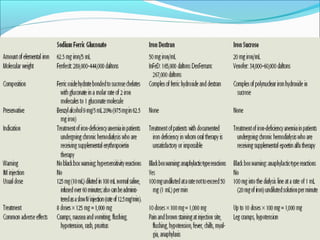
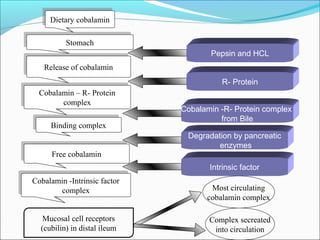
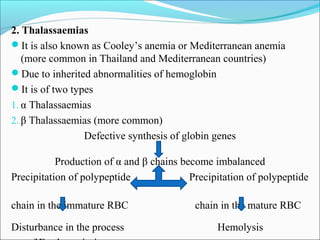
- Vitamin B12 deficiency can result from inadequate intake, malabsorption, or inadequate utilization. The major causes are pernicious anemia resulting from gastric acid and intrinsic factor deficiencies impairing vitamin B12 absorption.
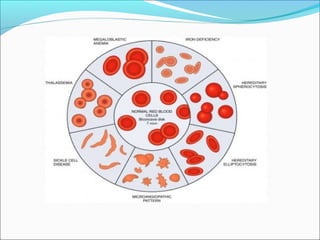
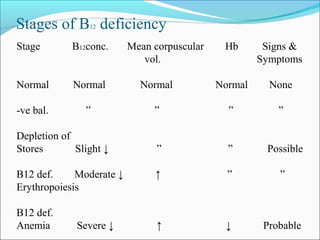
- There are three stages of vitamin B12 deficiency: stores depletion, impaired erythropoiesis, and anemia with neurological symptoms potentially developing at severe deficiency.
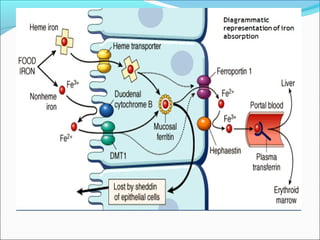
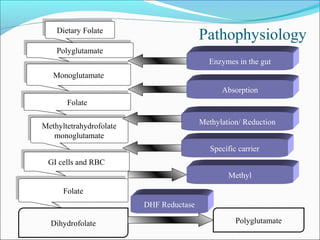
- Vitamin B12 works with folate in DNA/RNA synthesis and its absorption requires intrinsic factor to form a complex for distal ileum absorption. G








































































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)














