Downloaded 16 times







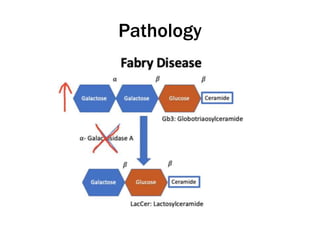

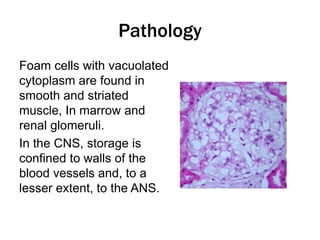


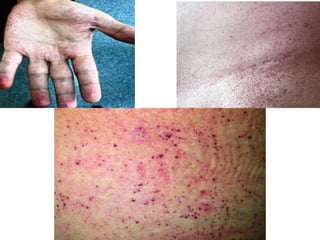

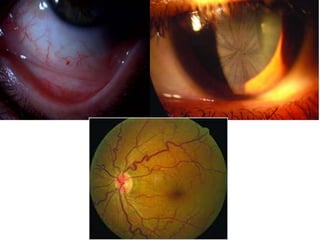
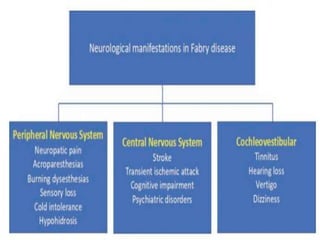







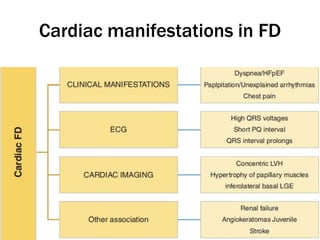



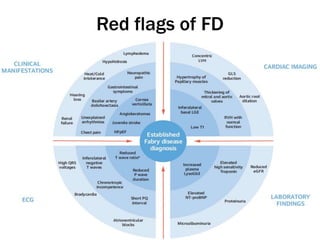





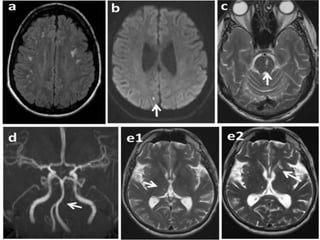











Fabry disease is a rare genetic disorder caused by deficient activity of the enzyme alpha-galactosidase A. This results in accumulation of globotriaosylceramide and related molecules in the body's cells. It is an X-linked recessive condition affecting males more severely than females. Symptoms involve the skin, eyes, kidneys, heart, and nervous system. Diagnosis is confirmed through enzyme testing or genetic analysis. Treatment includes enzyme replacement therapy with agalsidase beta, agalsidase alfa, or pegunigalsidase alfa. Migalastat is a pharmacological chaperone drug that can also be used. Management of organ-specific complications is also important.







































































![PERI-PROSTHETIC FRACTURE NAIL-PLATE CONSTRUCT [NPC].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/drarunkumardrmohamedashrafperiprostheticfrasturenail-plateconstructnpc-260209164459-7e9d15a1-thumbnail.jpg?width=640&height=640&fit=bounds)











![ONFH[AVN HIP] -TRIPLE REGIME -A NOVAL SURGICAL CONCEPT .pptx](https://cdn.slidesharecdn.com/ss_thumbnails/onfhavnhip2026koaconcalicutdrgokuldevdrmashraf-260210064517-213ec005-thumbnail.jpg?width=640&height=640&fit=bounds)



