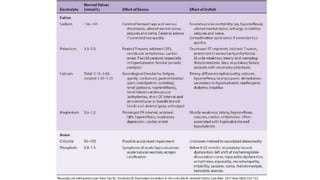
The document provides a comprehensive overview of electrolyte imbalances, focusing on potassium, calcium, phosphorus, and magnesium. It elaborates on the causes, clinical manifestations, and treatment strategies for conditions such as hypokalemia, hyperkalemia, hyponatremia, and hypercalcemia, highlighting the importance of electrolyte homeostasis in various health conditions. The document emphasizes specific management protocols, including oral and intravenous replacements, to mitigate acute risks associated with severe imbalances.



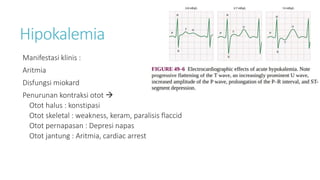
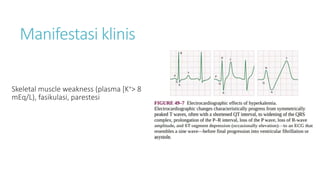
![Hipokalemia
[K+] < 3,5 mEq/L, karena :
1. Intercompartmental shift kalium: terapi insulin, alkalosis, hipotermia, ↑uptake K
dari eritrosit pada pengobatan asam folat/B12 pada anemia megaloblastik
2. ↑ Kehilangan kalium : peningkatan aktivitas mineralocorticoid, renal tubular
acidosis, ketoacidosis, amphotericin B, muntah/diare persisten, Keringat berlebih (jika
intake K+ kurang),
3. Intake kalium inadekuat](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-5-320.jpg)






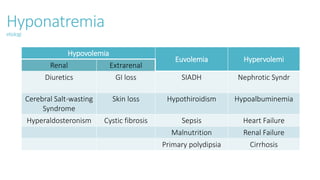
![Hyponatremia
[Na+] > 125 Tidak menunjukkan gejala neurologis
Tanda awal :
- Anoreksia
- Mual
- Kelemahan tubuh](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-15-320.jpg)



![Calcium
Normal plasma calcium concentration : 8.5 to 10.5 mg/dL (2.1–2.6 mmol/L)
50% is in the free, ionized form, 40% is protein bound (mainly to albumin), and 10% is
complexed with anions such as citrate and amino acids.
The free, ionized calcium concentration ([Ca 2+]) is physiologically most important. Plasma
[Ca 2+ ] is normally 4.75 to 5.3 mg/dL (1.19–1.33 mmol/L).
Tiap ↑/↓ 1g/dL albumin total plasma calcium concentration ↑/↓ 0.8-1 mg/dL
Tiap ↑/↓ pH 0,1 Ca 2+ ↑/↓ 0.16 mg/dL](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-19-320.jpg)
![Hypercalcemia
In primary hyperparathyroidism, secretion of PTH
is inappropriately increased in relation to [Ca 2+
].
In contrast, in secondary hyperparathyroidism
(eg, chronic kidney failure or malabsorption
syndromes), PTH levels are elevated in response
to chronic hypocalcemia.
Prolonged secondary hyperparathyroidism,
however, can occasionally result in autonomous
secretion of PTH, resulting in a normal or
elevated [Ca 2+ ] (tertiary hyperparathyroidism).](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-20-320.jpg)










![Hyperphosphatemia
Clinical manifestation :
Although hyperphosphatemia per se does not appear to be directly responsible for
any functional disturbances, significant hyperphosphatemia may produce
hypocalcemia via phosphate chelation with plasma [Ca 2+ ] and may also produce
acute kidney injury via parenchymal and tubular deposits of calcium-phosphate salts.
Hyperphosphatemia is associated with increased mortality in chronic kidney disease
and kidney failure patients, and is managed in this patient population by dietary
restriction, the use of phosphate binders, dialysis, or a combination of these
methods.
Treatment:
Hyperphosphatemia is generally treated with phosphate-binding antacids such as
aluminum hydroxide or aluminum carbonate.](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-31-320.jpg)




![Magnesium
Plasma [Mg 2+ ] is closely regulated between 1.7 and 2.1 mEq/L (0.7–1 mmol/L or 1.7–2.4
mg/dL) through interaction of the gastrointestinal tract (absorption), bone (storage), and
the kidneys (excretion).
Approximately 50% to 60% of plasma magnesium is unbound and diffusible](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-36-320.jpg)
![Hypermagnesemia
Increases in plasma [Mg 2+ ] are nearly always due to excessive intake (magnesium-
containing antacids or laxatives: magnesium hydroxide, Milk of Magnesia), kidney
impairment (GFR <30,L/min), or both
Less common causes include adrenal insufficiency, hypothyroidism, rhabdomyolysis, and
lithium administration. Magnesium sulfate therapy for preeclampsia and eclampsia can
result in maternal and fetal hypermagnesemia.](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-37-320.jpg)

![Treatment
Discontinue source(s) of magnesium intake (usually antacid or laxa
In cases of relatively high [Mg 2+ ] OR presence of clinical signs of magnesium toxicity,
intravenous calcium can temporarily antagonize most of the effects of clinical toxicity.
Forced diuresis with a loop diuretic and intravenous fluid replacement enhances urinary
magnesium excretion in patients with adequate renal function.
Dialysis will be necessary in such patients with significant kidney impairment or kidney
failure. Ventilatory or circulatory support, or both, may be necessary)](https://image.slidesharecdn.com/electrolyteimbalance-240620011742-482bdf03/85/Electrolyte-Imbalance-slide-explanations-39-320.jpg)