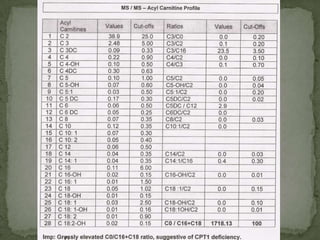
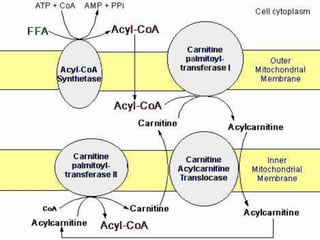
This document describes the case of a 3-year-old boy who presented with recurrent loss of consciousness following trivial illness. Initial workup revealed hypoglycemia and hyperammonemia. Further testing found elevated 2-oxoglutaric acid and a high C0/C16-18 ratio suggestive of carnitine palmitoyltransferase I (CPT-I) deficiency. The patient was diagnosed with a fatty acid oxidation disorder and treated accordingly.















































































































![CASE_PRESENTATION_ON_subdural_hematoma(SDH)[1 FINAL PPT]-1.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationonsubduralhematomasdh1finalppt-1-260129172522-d405d375-thumbnail.jpg?width=640&height=640&fit=bounds)












