

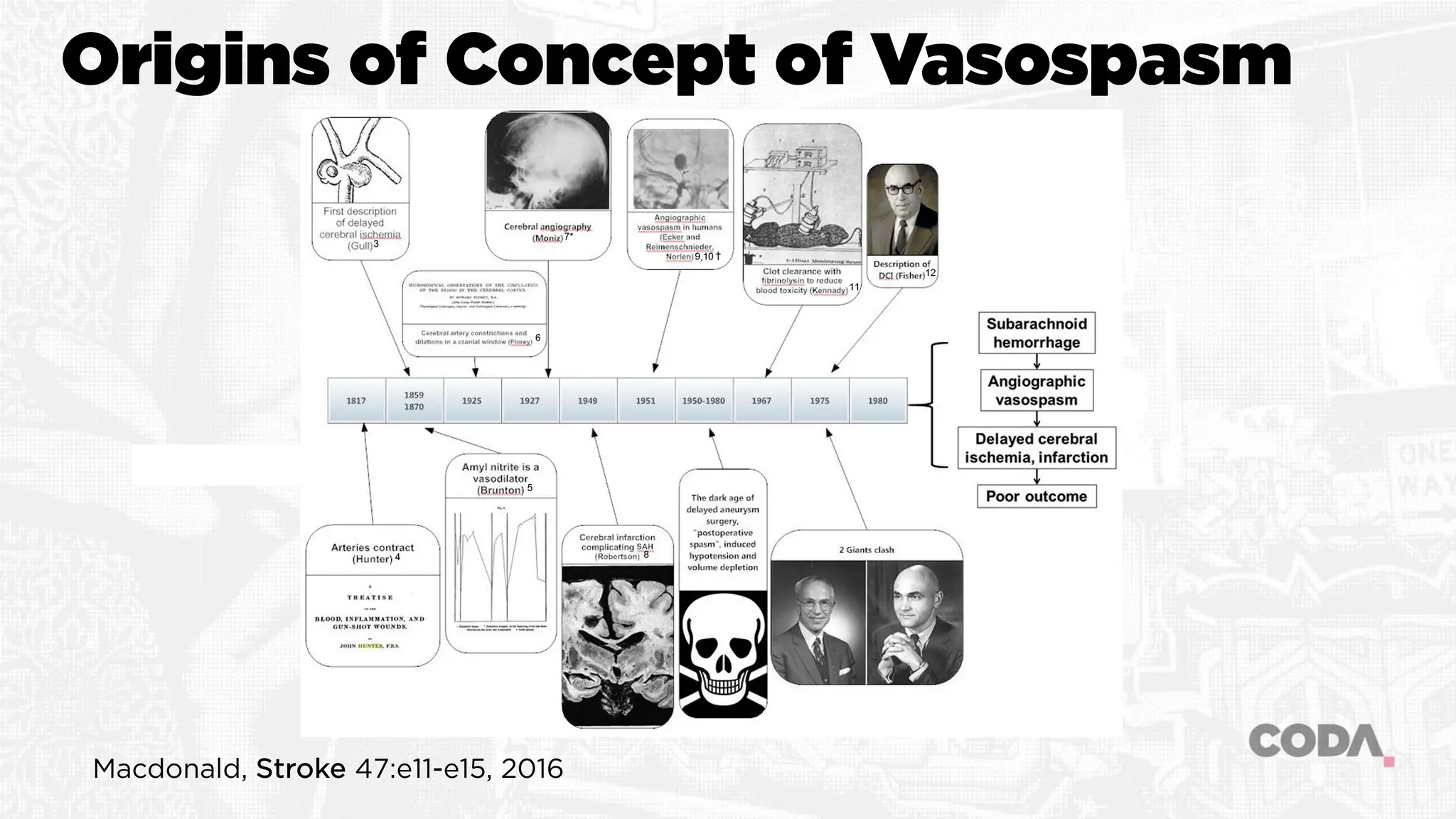
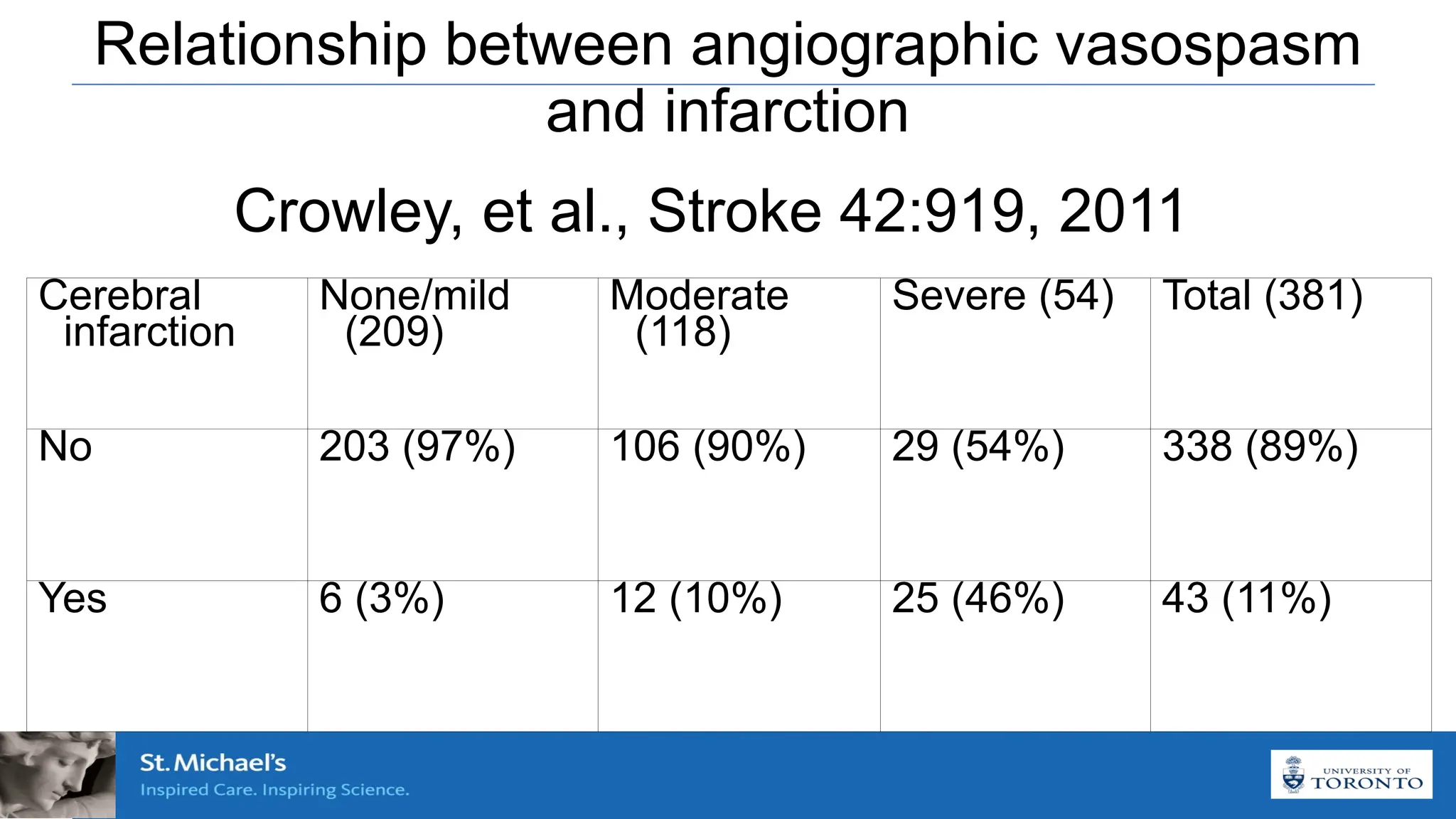
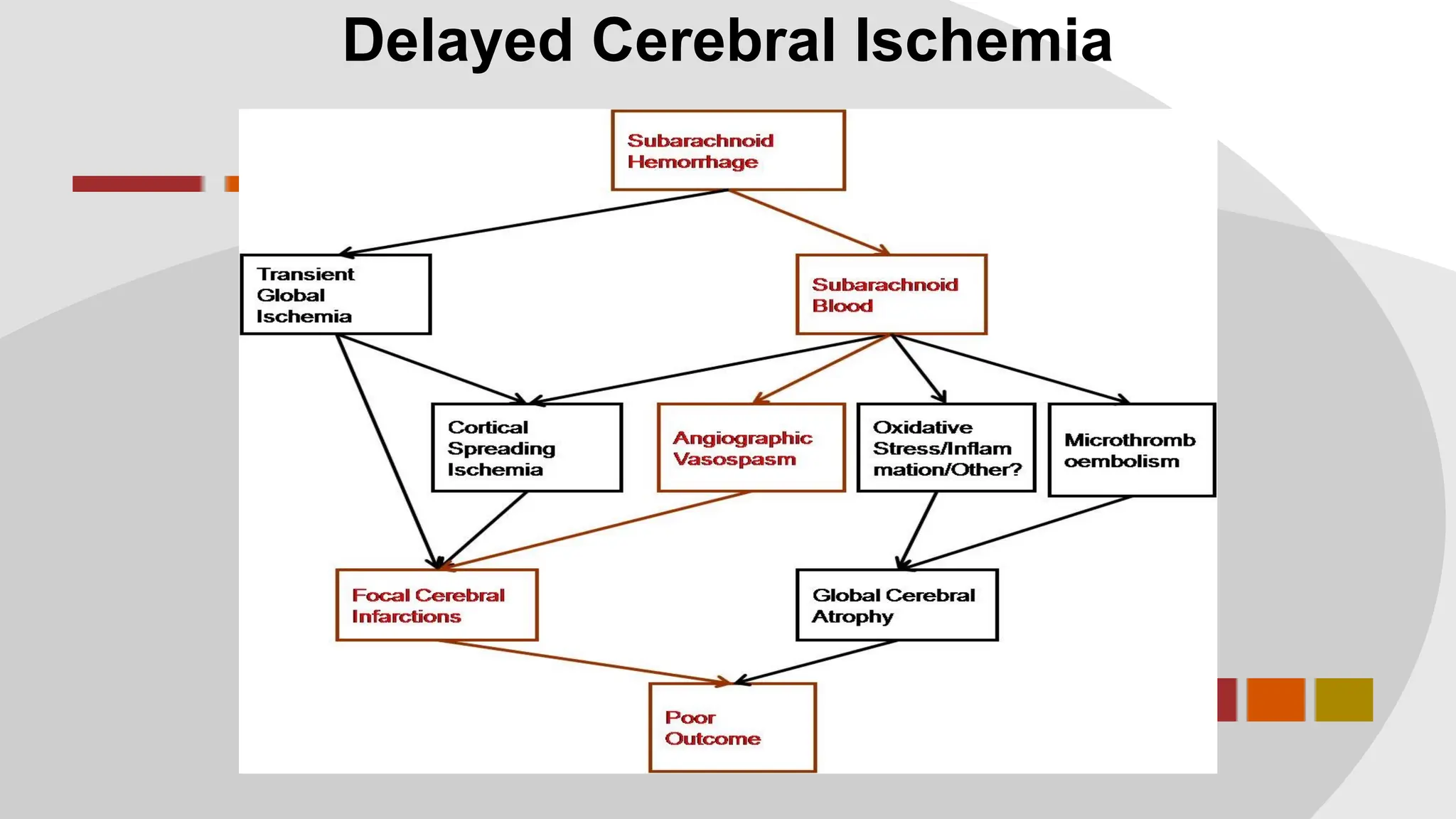
![Angiographic VSP
Blood clot leads to angiographic VSP leads to infarction and
poor outcome
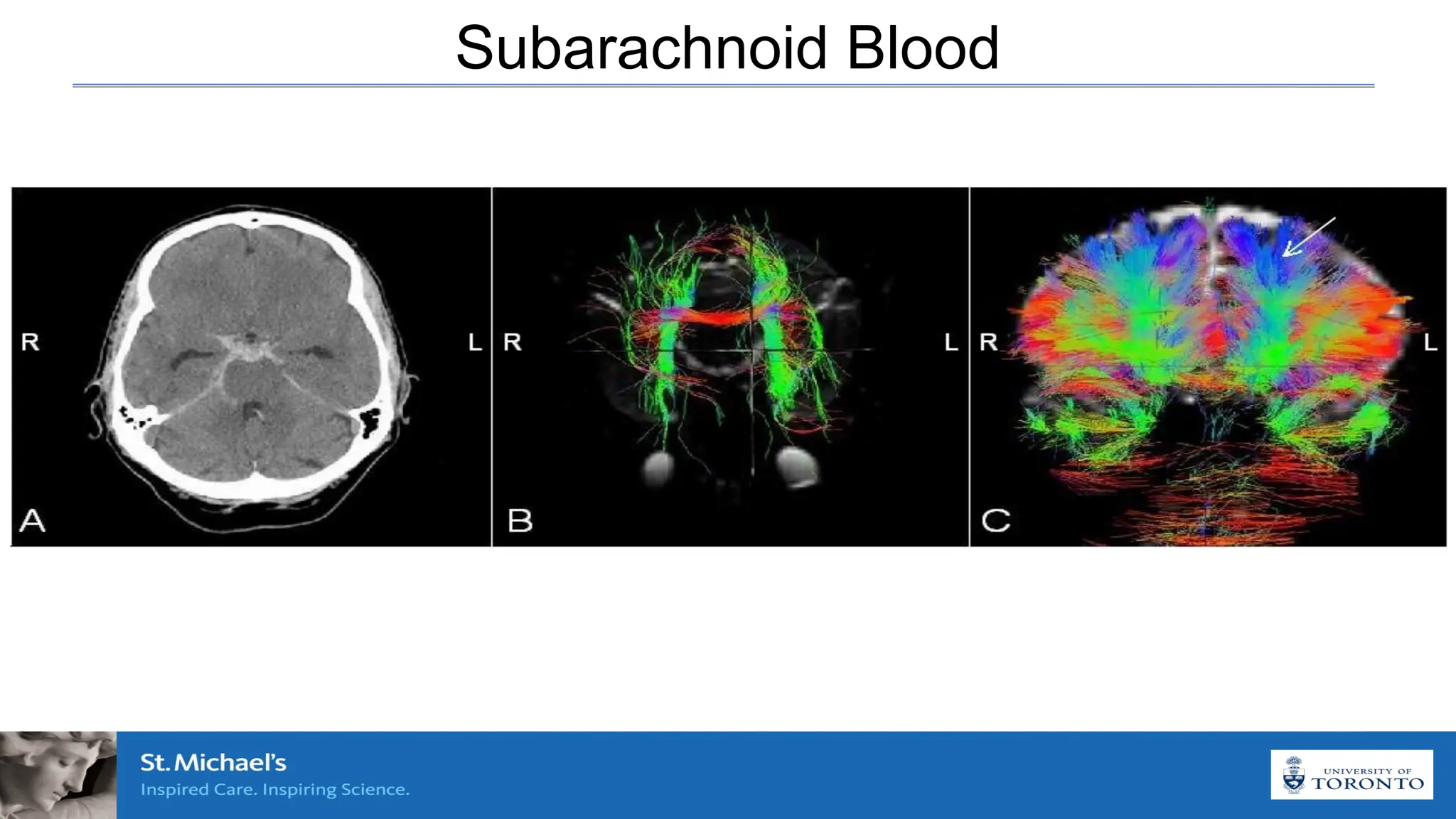
Vasoconstriction of large cerebral arteries due to hemoglobin
component of subarachnoid blood
“Pharmacologic” dose response related to the location,
duration of presence and concentration of the
hemoglobin/blood clot
Subarachnoid blood leads to angiographic vasospasm (?leads
to cerebral infarction [DCI] leading to poor outcome)](https://image.slidesharecdn.com/rlmcodavsptalkb-231201005705-b39f3231/75/Dilating-the-Dogma-of-Vasospasm-7-2048.jpg)

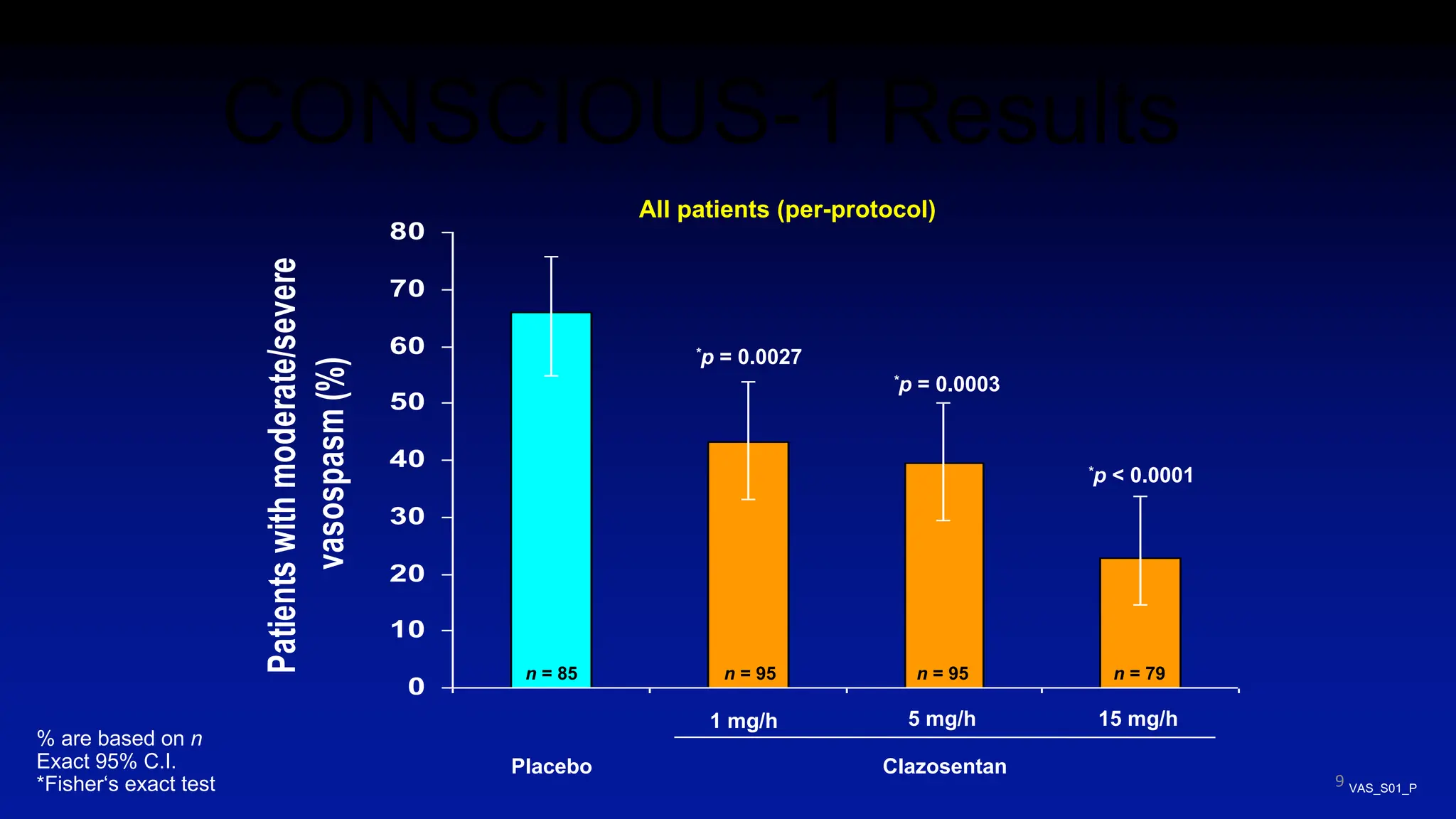
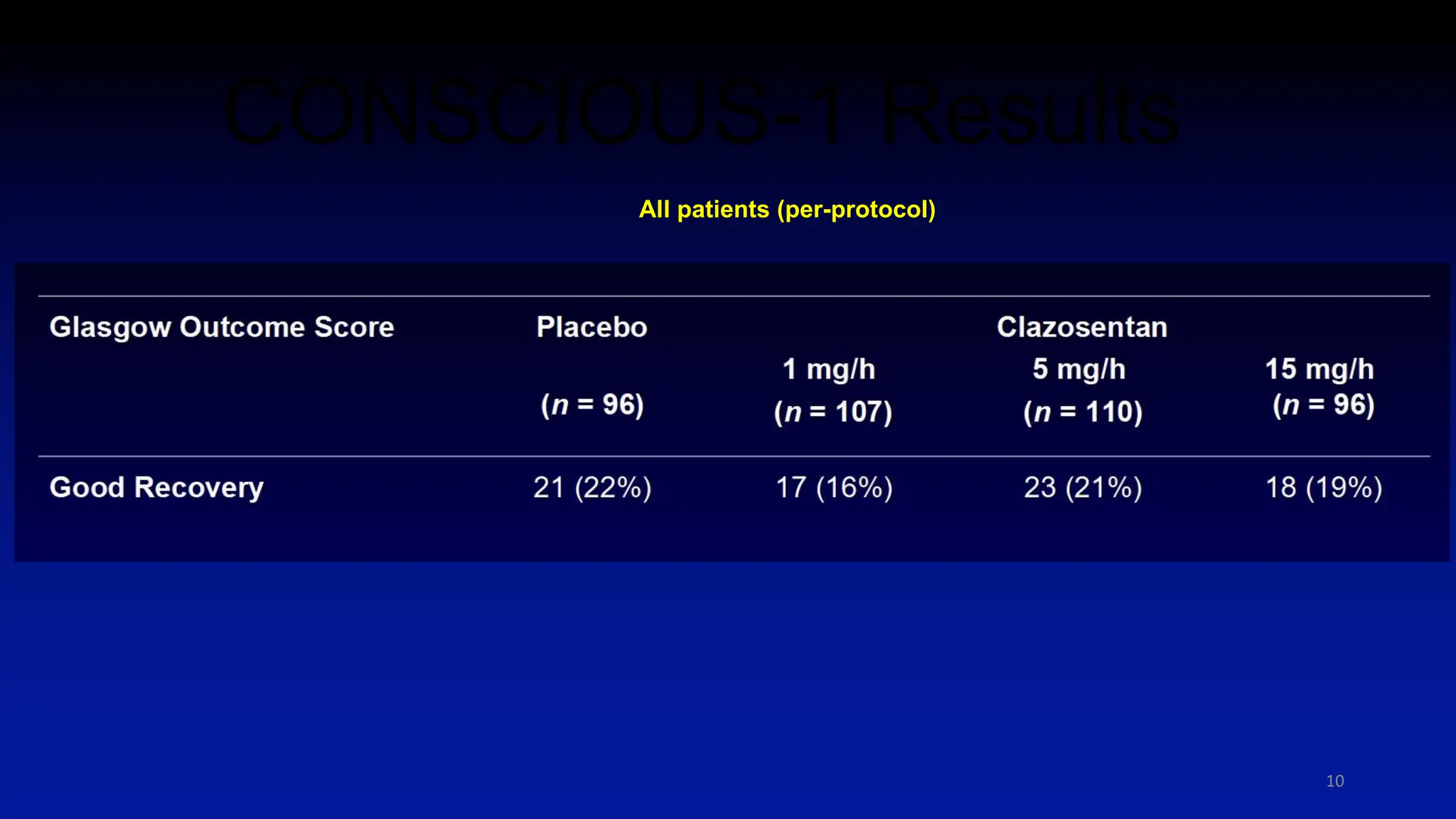












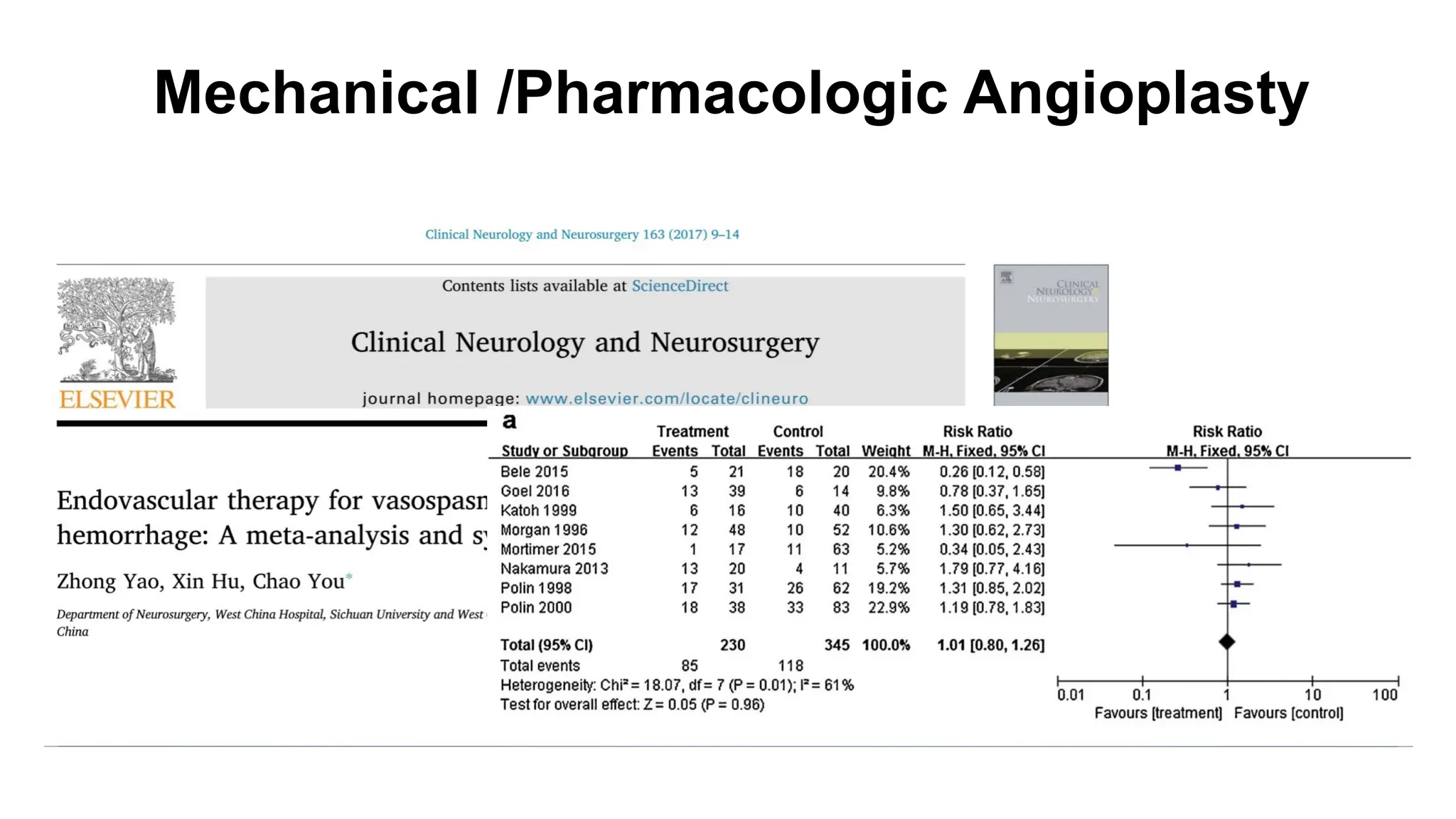

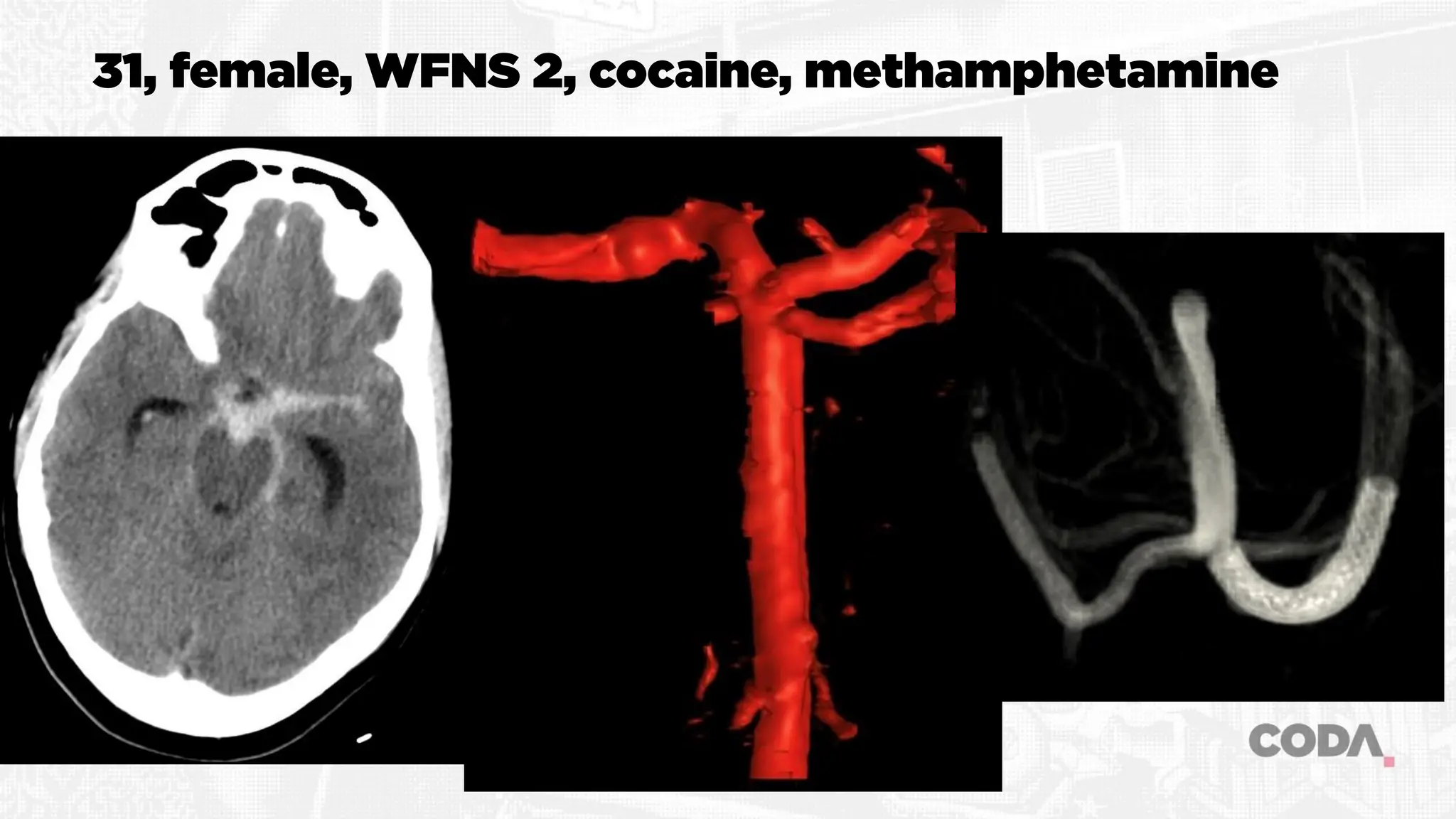
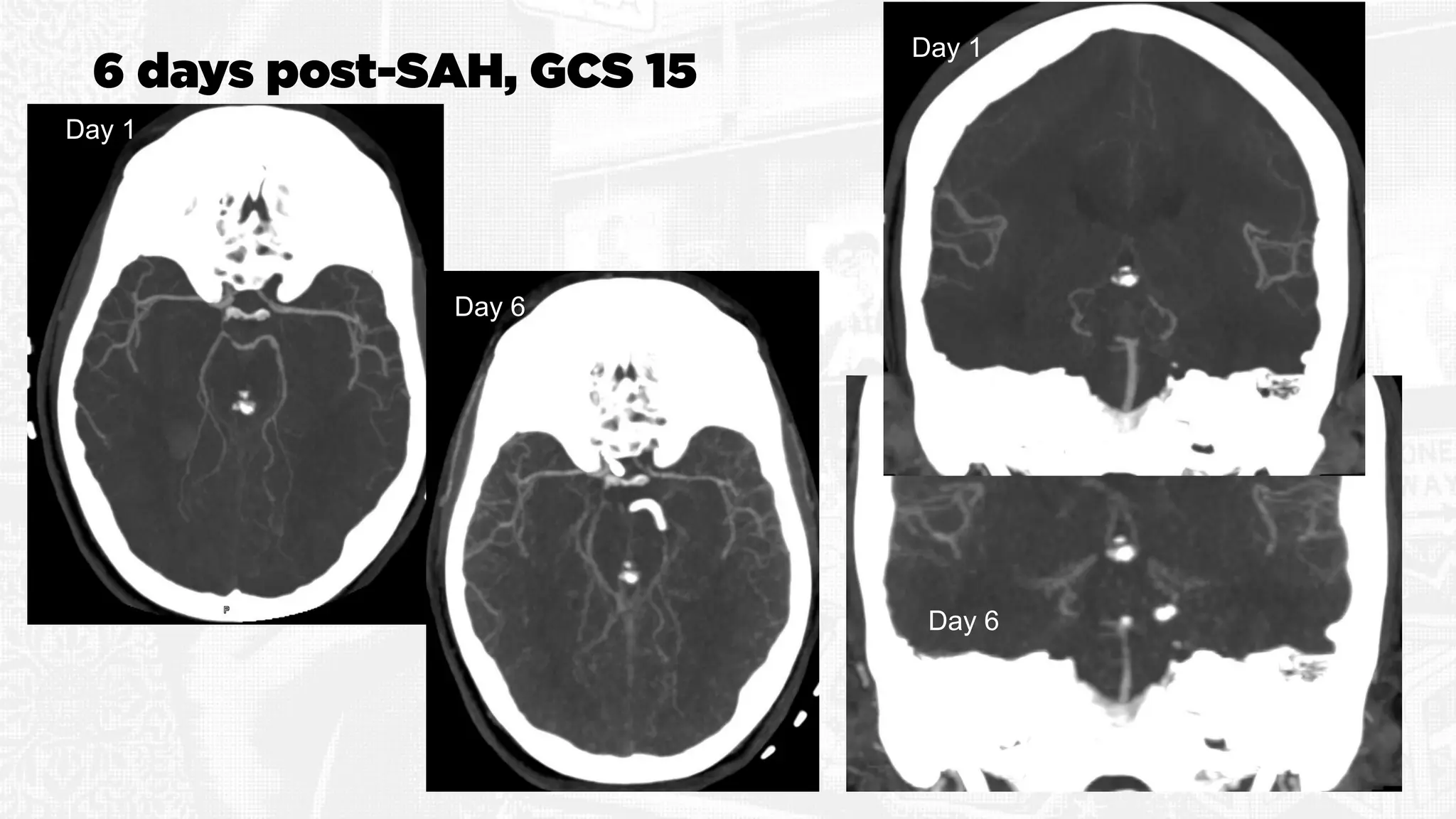
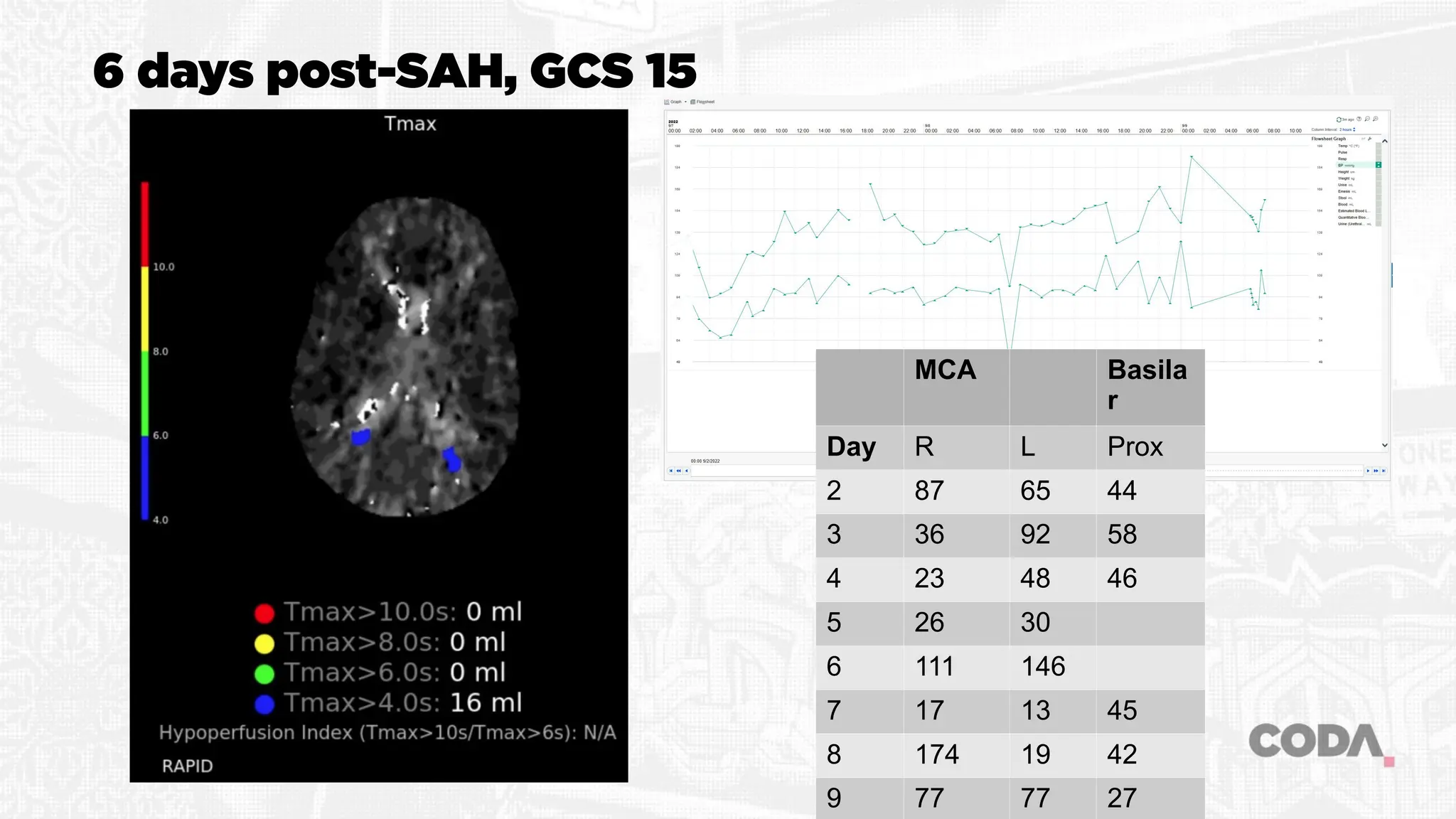
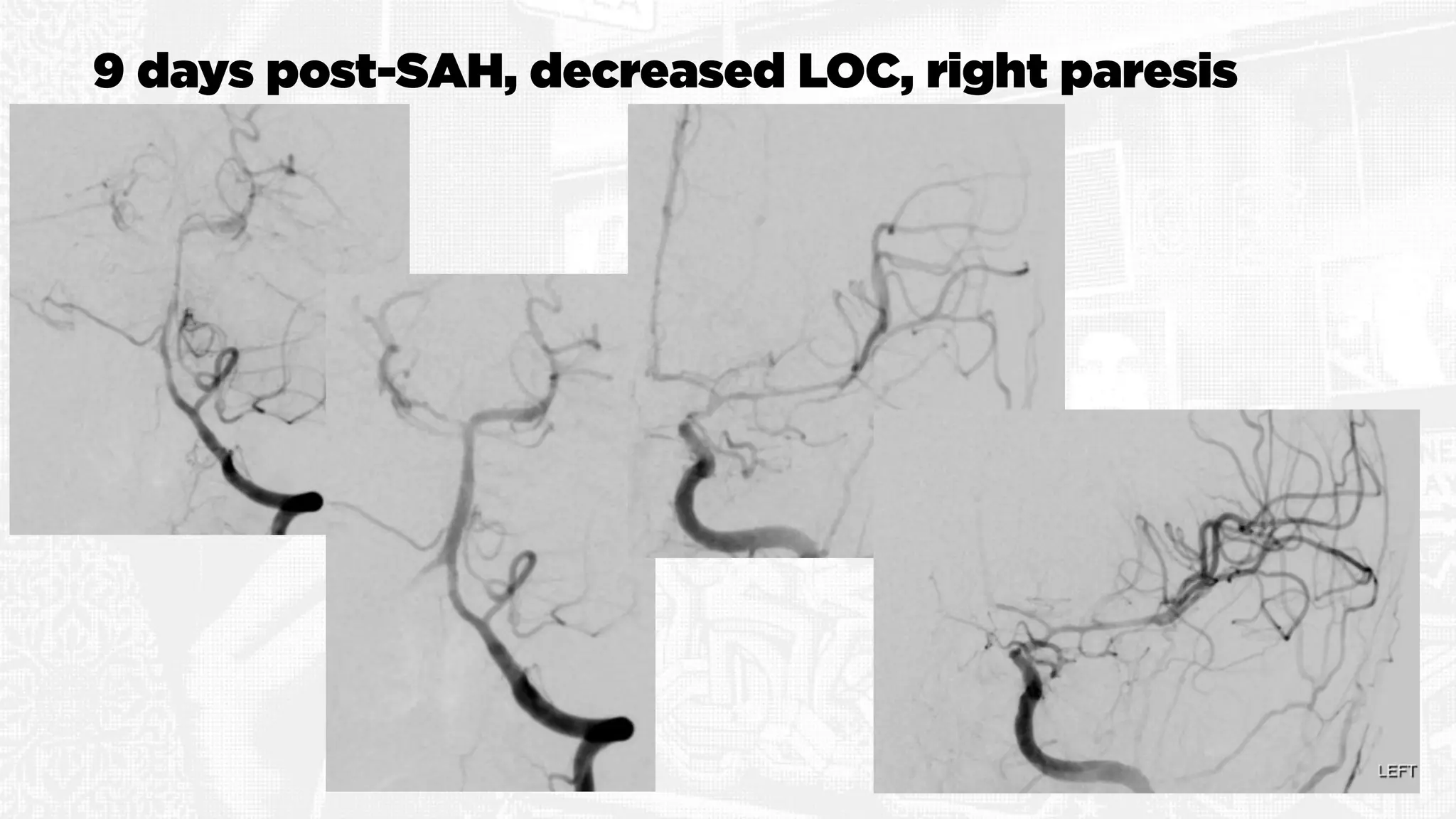
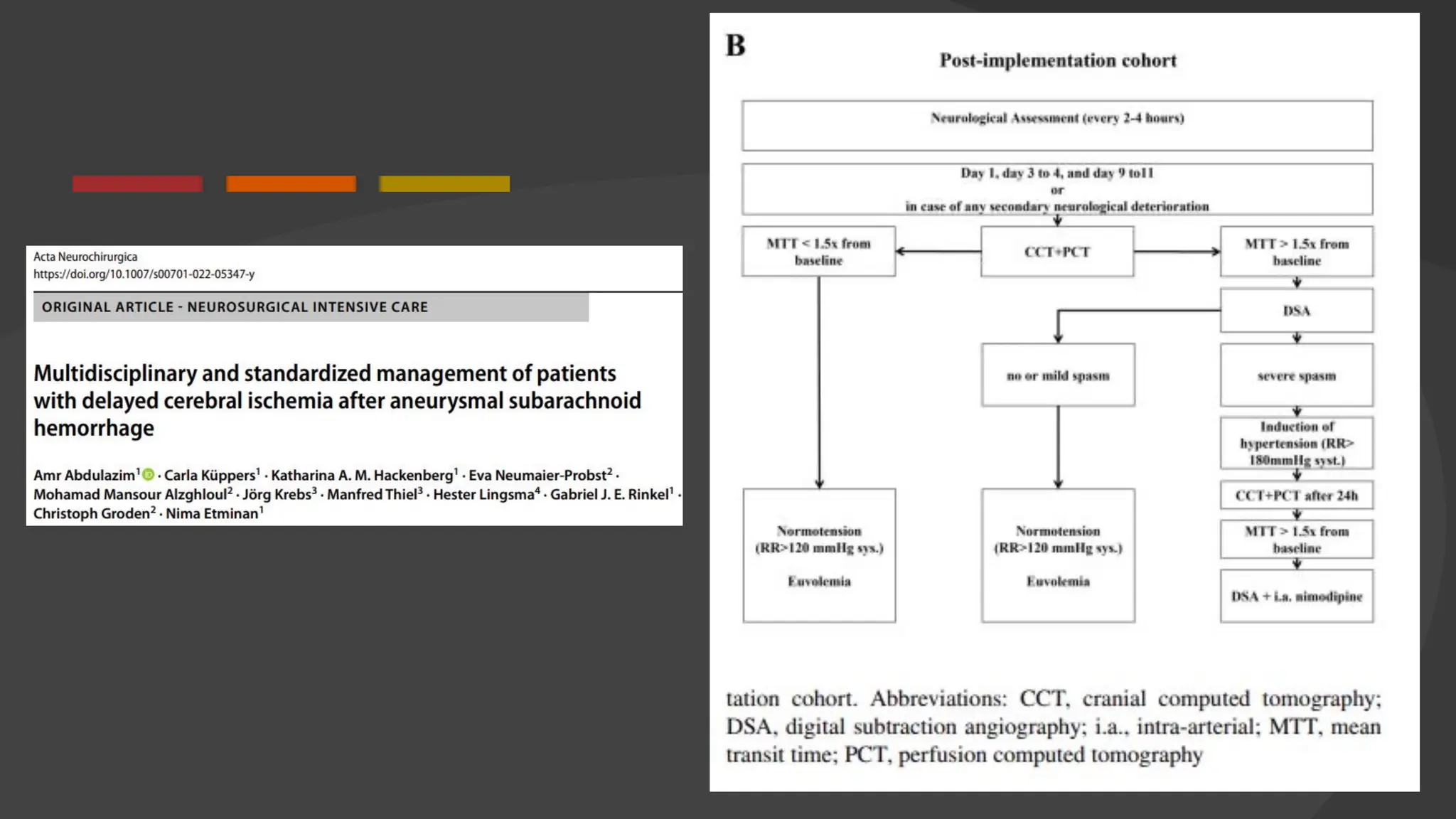




The document discusses the concept of vasospasm in relation to delayed cerebral ischemia (DCI) following subarachnoid hemorrhage (SAH), emphasizing the complexities surrounding its management and treatment outcomes. It outlines definitions, associated risks, and clinical guidelines for SAH management, including the use of intravenous nimodipine and the effects of induced hypertension. Additionally, it examines the challenges in establishing a direct relationship between angiographic vasospasm and poor clinical outcomes.























































![Apporach to lung biopsy [Auto-saved].pptx latest](https://cdn.slidesharecdn.com/ss_thumbnails/apporachtolungbiopsyauto-saved-251211225655-93258539-thumbnail.jpg?width=640&height=640&fit=bounds)










