This document provides summaries and introductions for the Dictionary of Pharmaceutical Medicine. It discusses the need for the dictionary to define terms, abbreviations, and acronyms used in the multidisciplinary field of pharmaceutical medicine. It acknowledges the dynamic nature of the field and challenges of keeping definitions up to date. The document contains forewords and prefaces from various experts in pharmaceutical medicine endorsing the dictionary as a useful reference for various professionals working in drug development.




































![27
cle
Times. March 15, 1995:A3. (Encyclopedia Article) Sturgeon T. Sci-
ence fiction. In: Lorimer LT, editorial director; Cummings C, ed-
in-chief; Leish KW, managing ed. The Encyclopedia Americana.
Vol 24. International ed. Danbury, Conn: Grolier Incorporated;
1995:390–392. (Book Article or Chapter) James NE. Two sides of
paradise: the Eden myth according to Kirk and Spock. In: Palumbo
D, ed. Spectrum of the Fantastic. Westport, Conn: Greenwood;
1988:219–223. (ERIC Document) Fuss-Reineck M. Sibling Com-
munication in Star Trek: The Next Generation: Conflicts Between
Brothers. Miami, Fla: Annual Meeting of the Speech Communi-
cation Association; 1993. ERIC Document Reproduction Service
ED364932. (Website) Lynch T. DSN trials and tribble-ations review.
Psi Phi: Bradley’s Science Fiction Club Web site. 1996. Available at:
http://www.bradley.edu/campusorg/psiphi/DS9/ep/503r.htm.
Accessed October 8, 1997. (Journal Article on the Internet) McCoy
LH. Respiratory changes in Vulcans during pon farr. J Extr Med [se-
rial online]. 1999;47:237–247. Available at: http://infotrac.galegroup.
com/itweb/nysl_li_liu. Accessed April 7,1999; there are other com-
mon styles such as of the “International Committee of Medical
Journal Editors, Uniform Requirements for Manuscripts Submitted
to Biomedical Journals”
, example: Halpern SD, Ubel PA, Caplan AL.
Solid-organ transplantation in HIV-infected patients. N Engl J Med.
2002 Jul 25;347(4):284–7.; → see Havard style, Vancouver style.
clastogen Substance causing toxic effects upon genetic material
(chromosomes) of cells, inducing permanent and transmissible
damages with microscopically detectable structural alterations
of chromosomes; → see also aneugen, genotoxicity, toxicity
tests.
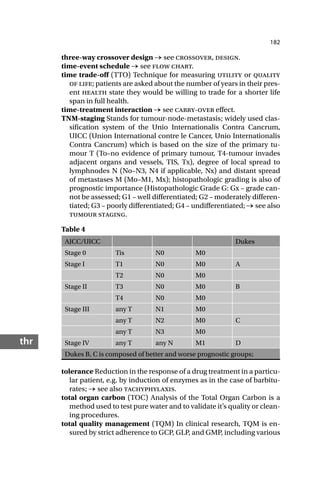
classification of recurrence Classification system used for describ-
ing recurrence of tumors after therapy: a = alive, without recur-
rence, B = alive with recurrence, C = alive, recurrence unknown,
D = dead without recurrence, E = dead with recurrence, F = dead,
recurrence unknown, G = lost, without recurrence; → see also tu-
mor staging.
clean area EC (IV): “an area with defined environmental control of
particulate and microbial contamination, constructed and used in
such a way as to reduce the introduction, generation and retention
of contaminants within the area”; → see also cross contamina-
tion.
clearance (Cl) rate of drug elimination from the body (volume of
blood cleared of a drug per minute: Cl = 0.693 (Vd
)/(t½) = ml/min)
where the volume of distribution (Vd
) is expressed in ml/kg and
the half-life (t½) in minutes or hours; → see also creatinine
clearance, elimination, pharmacokinetic.](https://image.slidesharecdn.com/dictionaryofpharmaceuticalmedicine1-231011045239-cd4dfff9/85/Dictionary_of_Pharmaceutical_Medicine-1-pdf-37-320.jpg)














![42
cos
are increasingly used to facilitate regulatory approval, justify pric-
ing and influence reimbursement; Australia and Canada request
economic data to support product application and reimbursement
listing since 1993, in France CEA and quality of life data are ex-
plicit criteria for determining prices and reimbursement; it is likely
that other European authorities will follow; → see also economic
analysis, effectiveness.
cost/minimisation analysis (CMA) Compares net costs of treat-
ments which have identical medical outcomes (patient outcomes
are all the same); not to be confounded with a cost-of-illness study,
where total costs (direct and indirect) attributable to a given illness
are calculated.
cost/utility analysis (CUA) Synthesizes simultaneously multiple
outcomes (e.g. on both morbidity and mortality, pain and phys-
ical function, but also quality) into a single measure; the basis for
this type of analysis is that each outcome is weighted by a person’s
preference (“utility”) for experiencing the outcome; CUA relates
therefore the costs of different procedures to the increased utility
which they produce e.g. in terms of quality-adjusted life-years
(QUALY) gained; the treatment with the lowest costs per QALY is to
be preferred; → see also utility measurement, quality of life.
Council for International Organisation of Medical Sciences
(CIOMS) International, non-governmental, non-profit organisa-
tion; set up under the auspices of the WHO and UNESCO; acts as
sounding board for capturing and disseminating informed opinion
on new developments in biology and medicine, but explores also
their social ethical, moral, administrative and legal implications;
well known is also the so called cioms I-form for reporting sus-
pect adverse reactions to the WHO centre in Uppsala and which
is accepted as report form by a number of health authorities, e.g.
in Germany, France, Italy, Ireland, The Netherlands and UK; this
form is almost identical with the form fda and accepted by
the US authority.
counterfeit medicine a medicine that is deliberately and fraudu-
lently mislabeled with respect to identity and/or content and/or
source.
covariate variable assumed to be related to the treatment re-
sponse.
creatinineclearance(CCr)Awidelyacceptedformulaforcalculating
the CCr from the serum creatinine Cr is that put forth by Cockcroft
and Gault (Nephron 1976, 16: 31–41): male CCr = (body weight [kg]
∞ (140 – age)) divided by (72 ∞ Cr [mg/100 ml]); female CCr = 0.85
∞ above; → see also clearance, glomerular filtration rate.
CRF correction log → see data resolution form.](https://image.slidesharecdn.com/dictionaryofpharmaceuticalmedicine1-231011045239-cd4dfff9/85/Dictionary_of_Pharmaceutical_Medicine-1-pdf-52-320.jpg)
































































![107
lot
phases between their bilayers; single-layered liposomes are gener-
ally < 0.1–0.2 µm in size and good carriers of water-soluble drugs;
their small size generally reduces their rate of elimination; multi-
layered vesicles range from about 1–5 µm; with a higher proportion
of lipid to aqueous phases due to multiple lipid bilayers, they are
suitable for transporting lipophilic drugs; they are more rapidly
cleared from the body than single-layered l.; → see also drug de-
livery systems, formulation.
listed adverse drug reaction ICH: “An adverse reaction whose
nature and severity are consistent with the information included
in the Company Core Safety Information; → see also unlisted
adverse drug reaction.
literature controls → see historical control.
loading dose syn. priming dose; initial dose of a drug which is
higher than the maintenance dose; the concept being to achieve
a therapeutic concentration more rapidly in case of drugs with a
slow elimination rate (e.g. therapy with tetracyclins, amiodarone,
digitalis glycosides).
local CRA → see clinical research associate.
local delivery → see route of administration.
log sheet Record of documents such as e.g. case record form, test ar-
ticle accountability form.
loi DMOS abbr. “Diverses Mesures d’Ordre Social”; French law con-
cerning financial benefits offered by the pharmaceutical industry
to physicians and all other members of medical professions.
loi Huriet syn. Loi Huriet-Serusclat; French Medicines Act which
came into operation in December 1988.
long-term use EC: “where the medicine is likely to be administered
regularly over a substantial period of life, i.e. continuously during
a minimum period of 6 months or frequently in an intermittant
manner so that the total exposure is similar”
.
Lorentz-formula For calculating the ideal body-weight (w) of a
subject; for men: w = (height [cm] – 100) – ((height – 150)/4); for
women: w = (height – 100) – ((height – 150)/2); → see also body-
mass-index.
loss to follow-up Patients lost to a clinical trial without knowing the
reasons; sometimes also used to describe the total number of pa-
tients lost, i.e. not finishing a particular clinical trial (premature
termination; the FDA (Clinical Guidance Document, 1994) sug-
gests that loss to follow-up should be less than 20%; → see also
drop-out, withdrawal.
lot FDA: “batch, or a specific identified portion of a batch, having
uniform character and quality within specified limits; or, in case
of a drug product produced by continuous process, it is a specific](https://image.slidesharecdn.com/dictionaryofpharmaceuticalmedicine1-231011045239-cd4dfff9/85/Dictionary_of_Pharmaceutical_Medicine-1-pdf-117-320.jpg)




















































![161
res
rescue medication also called escape medication; medicines identi-
fied in a study protocol as those that may be administered to the
patients when the efficacy of the investigational medicinal product
(IMP) is not satisfactory, or the effect of the IMP is too great and
is likely to cause a hazard (adverse reaction) to the patient, or to
manage an emergency situation).
research and development (R&D) The average new chemical en-
tity (NCE) costs about 802 million US $ (2002) (estim. 1990: $ 231
million) and takes about 12 years, with little changes since 1987,
from synthesis to marketing approval (about 3 years in the 1960s);
this includes costs of failed projects and time costs (mean develop-
ment times: long-term animal studies 3.5 years, phase I 1.5 years,
phase II 2 years, phase III 3.5 years, new drug application review
by FDA 1.5 years); to bring 10 NCEs on the market, it is estimated
that researchers must evaluate 100,000 compounds of which com-
panies can put about 100 products into clinical trials – but only
two of the 10 NCE will be profitable for the discovering company;
worldwide R&D expenses were about 49 billion US$ in 2004 (esti-
mate of the PhRMA) (after 15,000 million US $ in 1988, 24,500 mil-
lion in 1992 and 33,700 million in 1995), with about 15–20 NCEs
approved each year (35 in 1989); each additional week of clinical
development accounts for a loss of sales revenues in the order of
$ 1–10 million US $; worldwide ethical pharmaceutical sales were
in the order of $ 400,000 in 2002 (112.000 million in 1988); R&D ori-
ented companies expend about 10–20% of their revenues for R&D
and 20–30% for marketing; → see also development, health care
costs, life cycle management, new chemical entity, market-
ing authorisation, pharmaceutical market.
research coordinator → see clinical trial coordinator
research nurse → see study nurse.
response R. can be presented in different ways, e.g. as difference
(value before – value after), as ratio (value after/value before), as
percentage change [(value after/value before – 1) × 100], percentage
of patients with a defined value at a given moment, a.s.o.
response (cancer treatment) For reporting results of cancer treat-
ment the following definitions (WHO) of objective response are
used (separately!): (I) measurable disease: complete response (CR)
= disappearance of all known disease, determined by 2 observa-
tions not less than 4 weeks apart; partial r. (PR) = 50% or more
decrease in total tumour size of the lesions which have been mea-
sured to determine the effect of therapy by 2 observations not less
than 4 weeks apart (there can be no appearance of new lesions or
progression of any lesion); no change (NC) = 50% decrease in total
tumour size cannot be established nor has a 25% increase in the](https://image.slidesharecdn.com/dictionaryofpharmaceuticalmedicine1-231011045239-cd4dfff9/85/Dictionary_of_Pharmaceutical_Medicine-1-pdf-171-320.jpg)