Download as PDF, PPTX


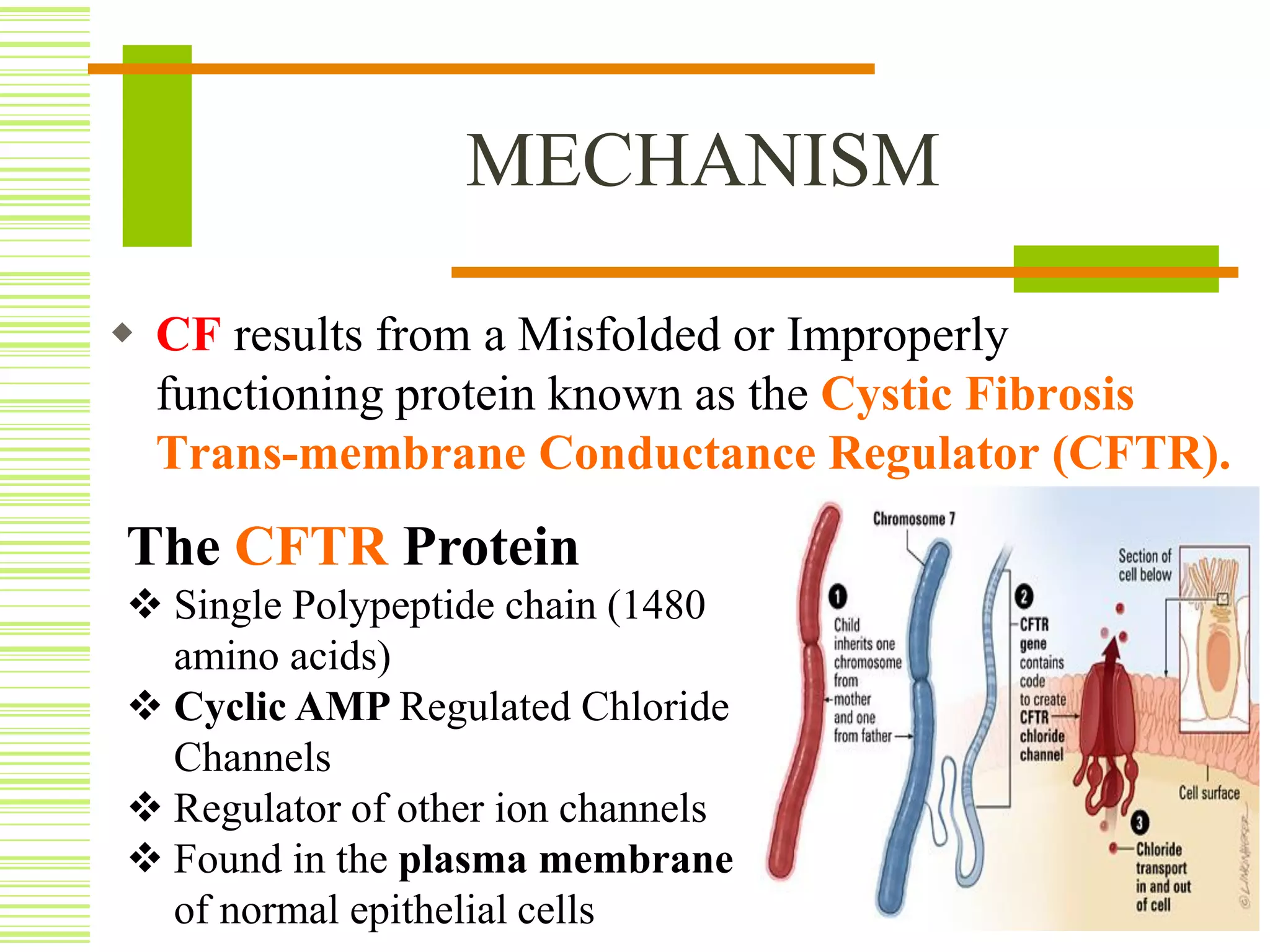
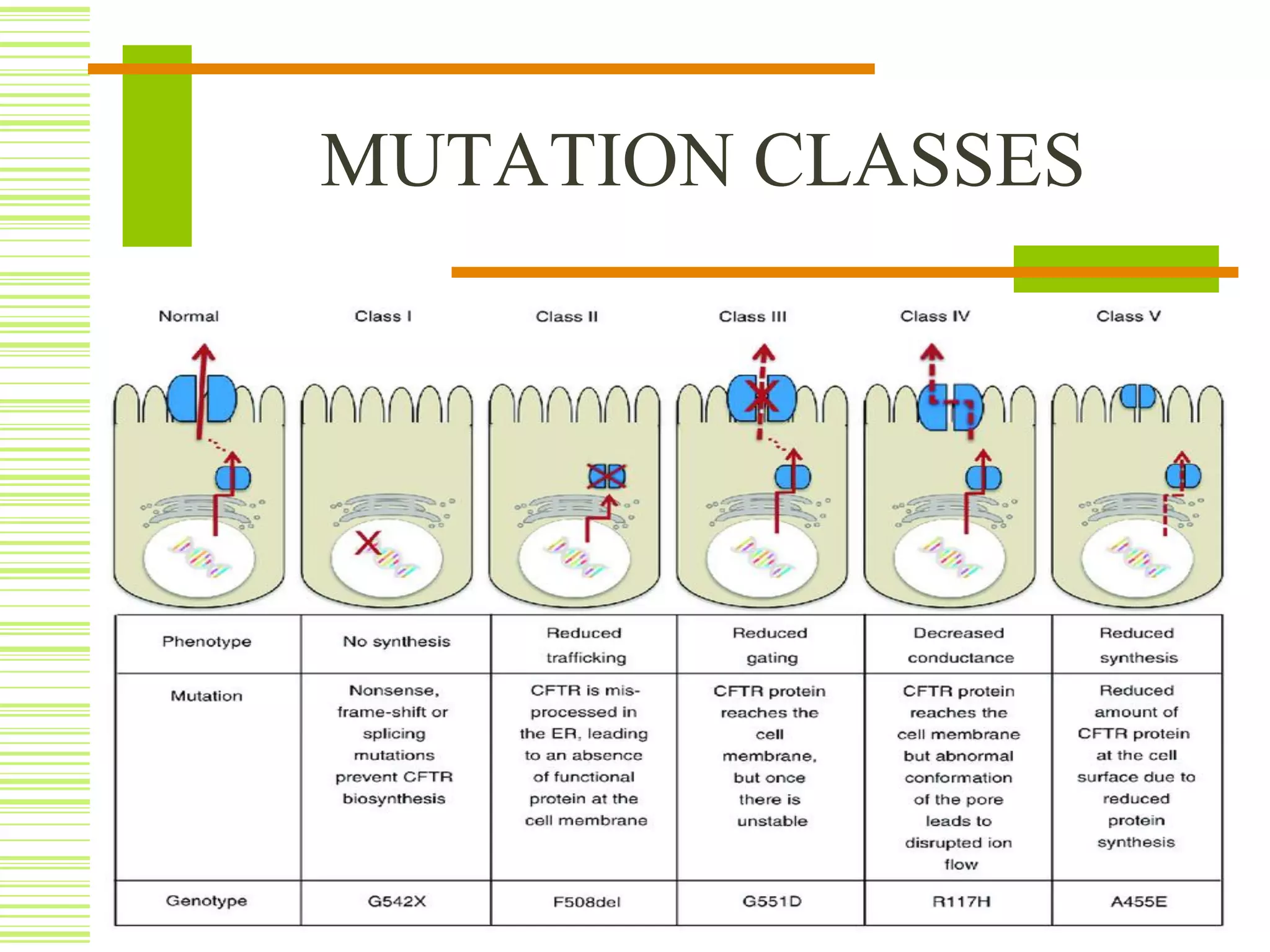










Cystic fibrosis is an inherited disorder that affects the cells that produce mucus, sweat and digestive juices. It is caused by a mutation in the CFTR gene that results in a misfolded CFTR protein. This prevents proper water flow and leads to thick, dehydrated mucus. The buildup of mucus in the lungs and pancreas can cause life-threatening lung infections and problems with digestion. Cystic fibrosis is typically diagnosed through newborn screening, genetic testing, sweat tests, imaging and lung function tests. While there is no cure, treatment focuses on loosening mucus, treating infections, improving nutrition and managing symptoms with medications like antibiotics, enzymes and airway clearance techniques.

![Cystic fibrosis[1]](https://cdn.slidesharecdn.com/ss_thumbnails/cysticfibrosis1-101115143757-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



























































