Download to read offline


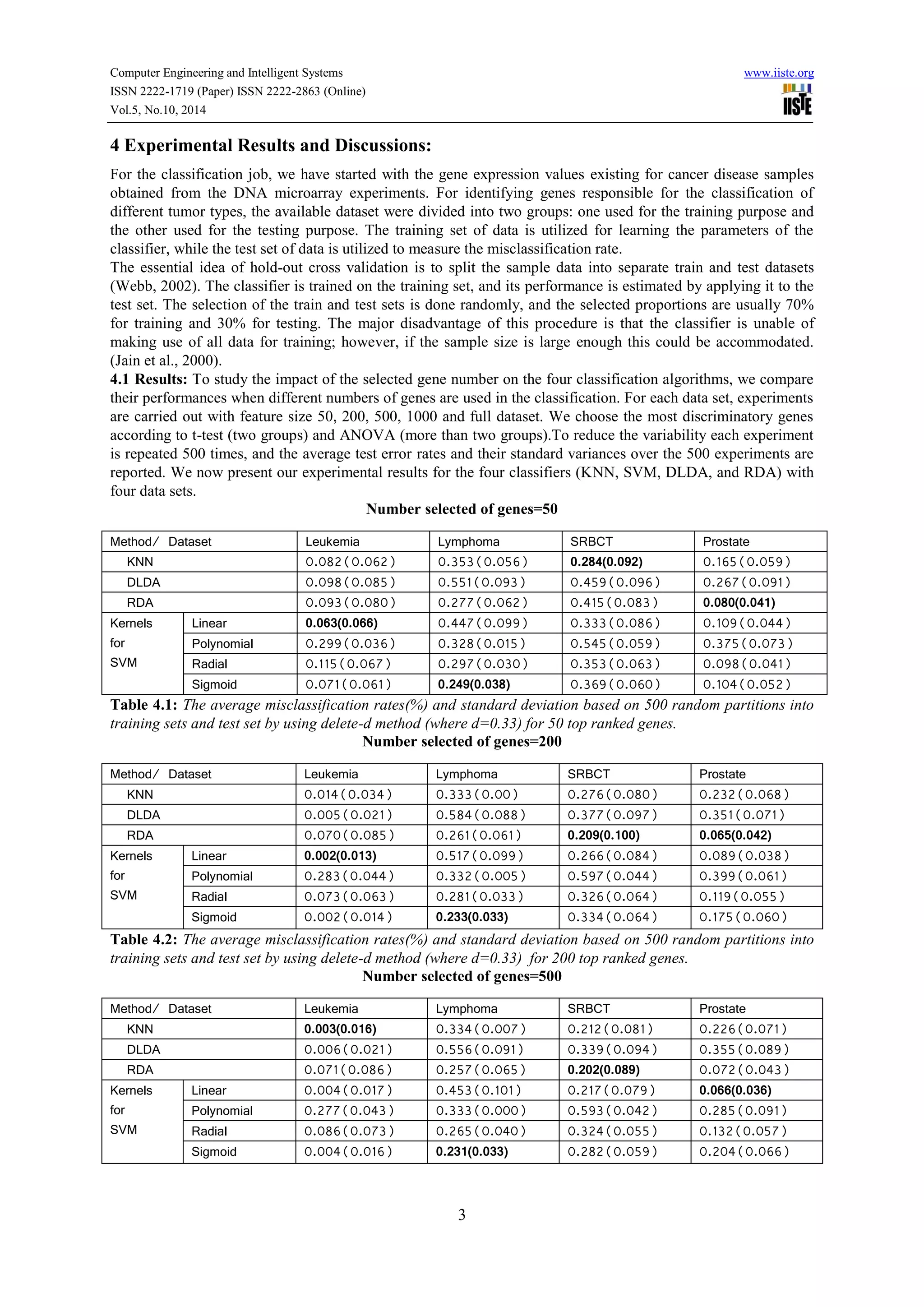
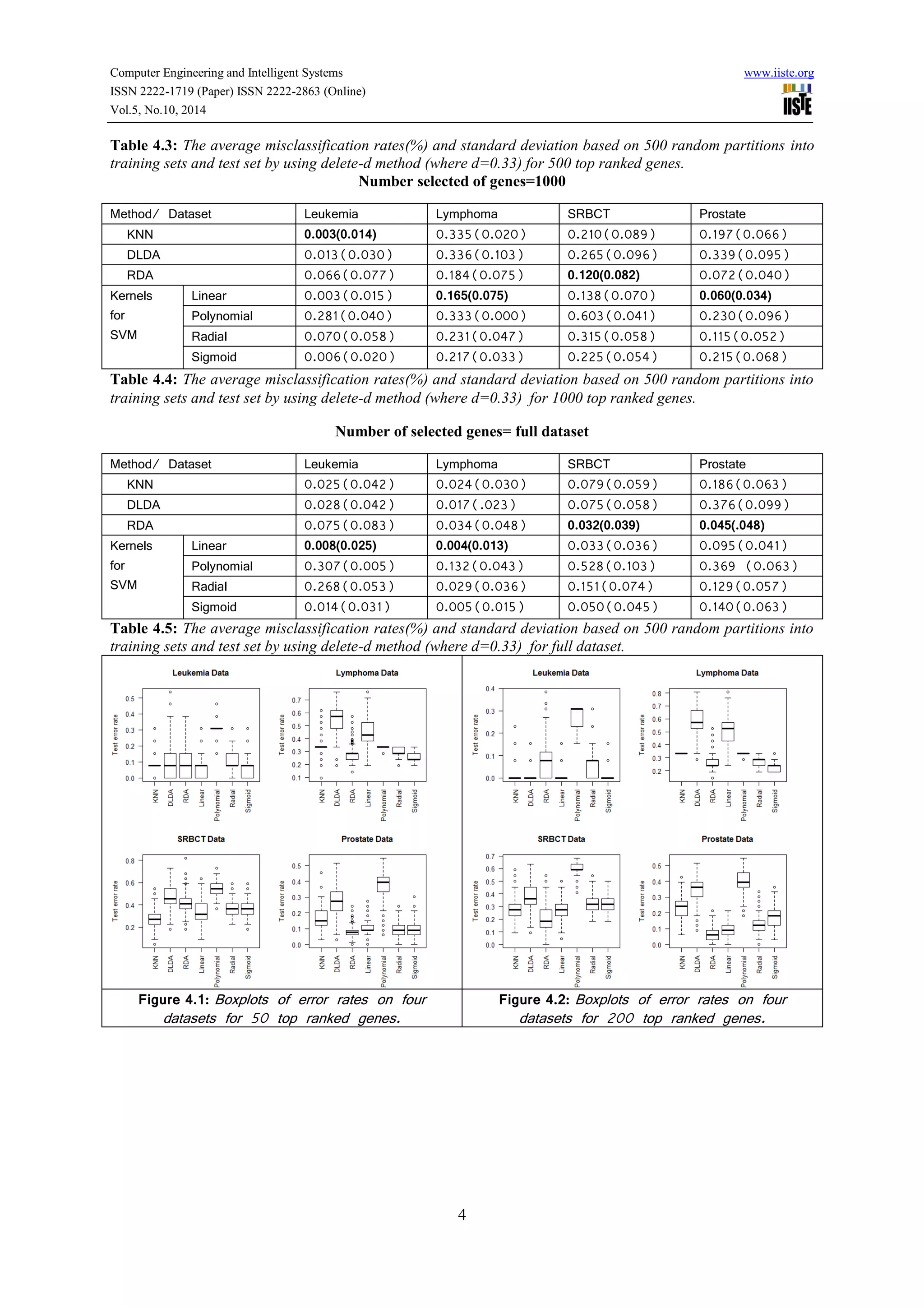

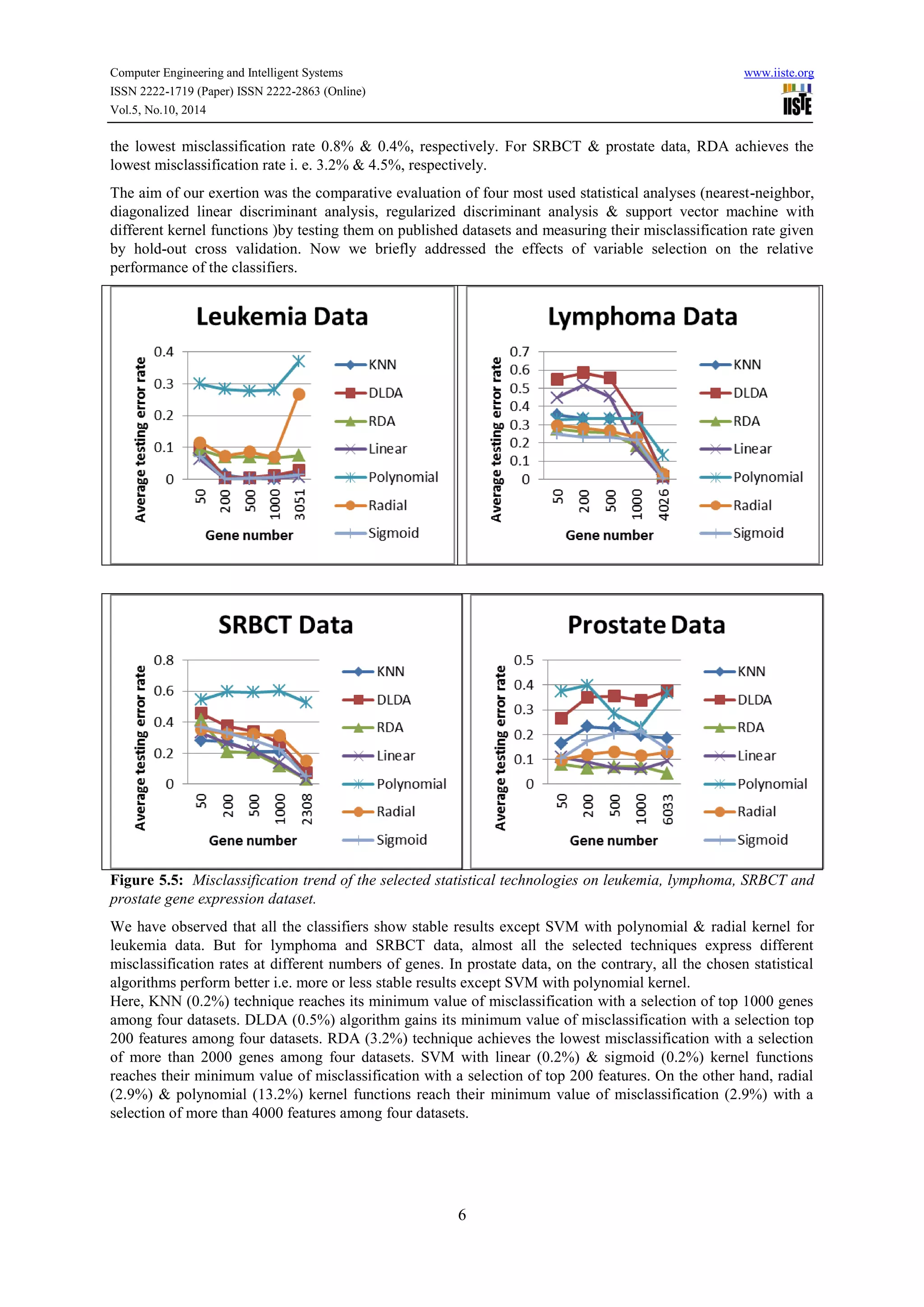





This document compares the predictive accuracy of various machine learning and statistical classification algorithms on four gene expression datasets. It finds that KNN, RDA, and SVM with a linear kernel generally have lower misclassification rates than DLDA when classifying tumors using different numbers of selected genes (50, 200, 500). Classification performance is evaluated using hold-out cross-validation, and the algorithms are tested on leukemia, lymphoma, SRBCT, and prostate cancer gene expression datasets containing between 38 and 102 samples each.
































































