


















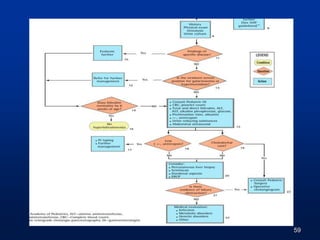
![EXAMINATION: [AH(R&R)]
07 months
Child is active, alert & oriented to mother
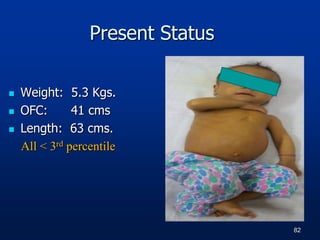
Weight- 5.000 Kg (7.450 kg) < 3rd percentile
OFC- 40 cm (44 cm) < 3rd percentile
Length-58 Cms (66.10 cms) < 3rd percentile
21](https://image.slidesharecdn.com/cholestasis8-130604121455-phpapp01/85/Cholestasis-8-21-320.jpg)
![ HR-108/min (90-120/min)
RR-30/min (30-40/min)
Temp-Afebrile
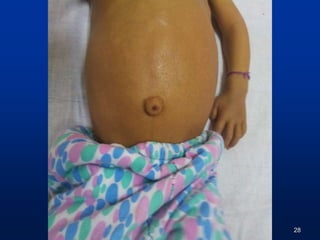
Icterus++ up to soles
No dysmorphic features
EXAMINATION: [AH(R&R)]
22](https://image.slidesharecdn.com/cholestasis8-130604121455-phpapp01/85/Cholestasis-8-22-320.jpg)











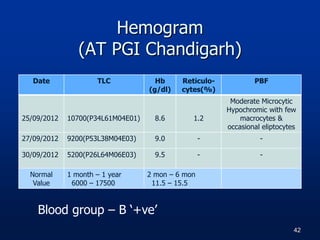
![Hemogram [AT AH(R&R)]
Date 27/01/13
Hb 11.3 gm/dl
TLC 8400cells/cmm P-28
L-57
PLT 2,30,000/cmm
Date 04/02/13
Hb 11.8 gm/dl
TLC 10,400cells/cmm P-84
L-10
PLT 2,03,00cells/cmm
Normal Value
TLC :1 mon – 1 yr
6000 - 17500
Hb : 11.5 – 15.5
g/dL
44](https://image.slidesharecdn.com/cholestasis8-130604121455-phpapp01/85/Cholestasis-8-44-320.jpg)

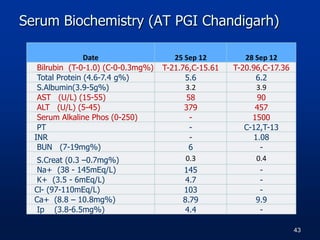
![Serum Biochemistry [AT AH(R&R)]
Date 27/01/2013 04/2/2013 15/02/2013
S. Bilrubin (T-0-1.0) (C-0-
0.3mg%)
T-17.7, C-10.5 T-31.2, C-22.0 T-21.8, C-11.7
ALT (15-55U/L) 349 1098 420
AST (U/L) (5-45) 114 425 116
Alkaline Phosphatase 1110 1387 1447
GGT (8-90IU/L) 48 - 36
Total Protein(4.6-7.4g%) 5.3 - 6.1
S.Albumin (3.9-5g%) 3.4 - 3.6
BUN (7-19mg%) 10 0.6 8
S.Creat (0.3 – 0.7mg%) 0.3 0.2 0.2
Na+ (138 - 145mEq/L) 139 136 140
K+ (mEq/L) (3.5 - 6) 4.2 5 4.4
46](https://image.slidesharecdn.com/cholestasis8-130604121455-phpapp01/85/Cholestasis-8-46-320.jpg)

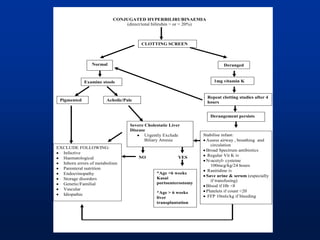
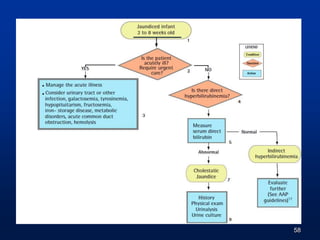
![Neonatal Cholestasis
Prolonged neonatal jaundice
[Jaundice that last longer than 14 days (term) or 21
days (premature babies)]
Conjugated hyperbilirubinemia
Conjugated bilirubin >1mg/dL if TB < 5mg/dL
>20% Total Bilirubin if TB is >5 mg/dL
High coloured urine with or without acholic stools
Occurs in 1:2500 live births
48](https://image.slidesharecdn.com/cholestasis8-130604121455-phpapp01/85/Cholestasis-8-48-320.jpg)
![Alpha 1 Antitrypsin
123 mg/dl (90.00-200.00)
[24/09/2012]
G6PD 2.00 U/g of Hb (>2.00)
Cystic Fibrosis (IRT) 21.40 ng/ml B (<70.00)
Other investigation
49](https://image.slidesharecdn.com/cholestasis8-130604121455-phpapp01/85/Cholestasis-8-49-320.jpg)


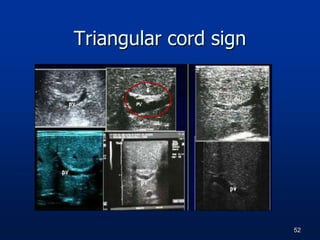
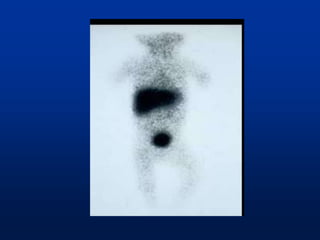
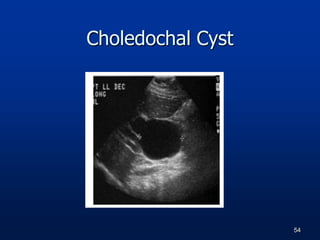



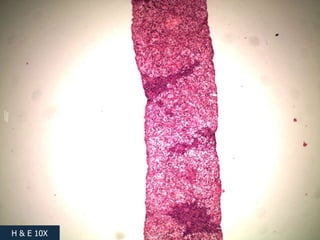
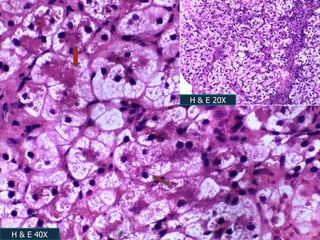
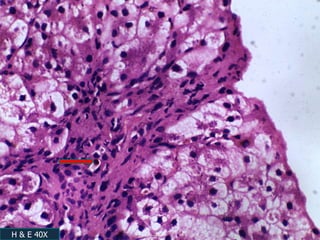
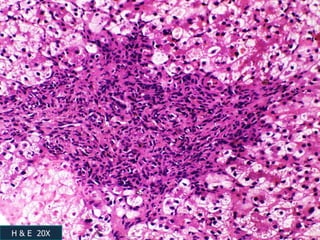
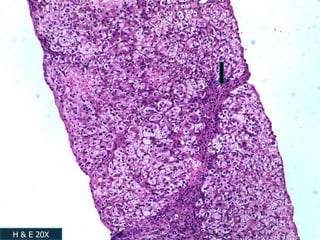
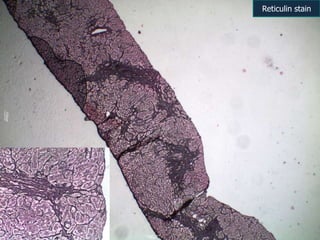
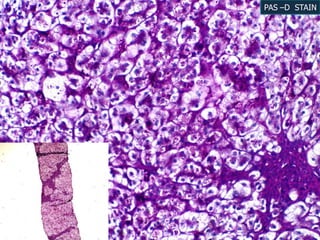
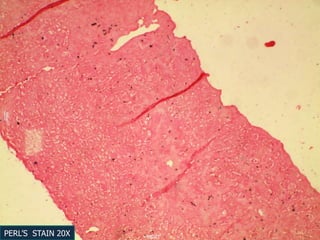









A 7 month old female infant presented with persistent jaundice since 2 weeks of life, high colored urine, and normal colored stools. On examination, she had deep icterus, hepatomegaly, and failure to thrive. Initial tests showed conjugated hyperbilirubinemia. Further workup included normal thyroid function, urine tests, TORCH titers, and alpha-1 antitrypsin level. Liver function tests showed elevated enzymes. Imaging showed hepatomegaly. Differentials included genetic and infectious causes of neonatal cholestasis.



































































