

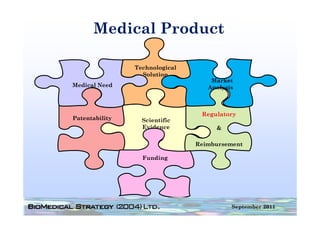



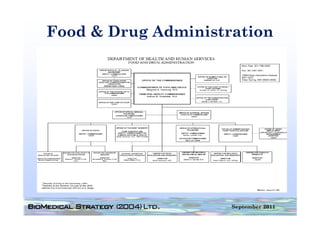
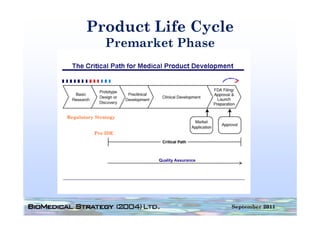

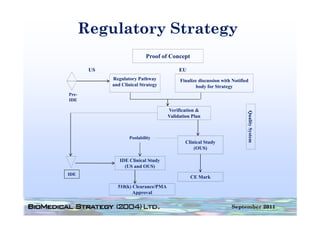



























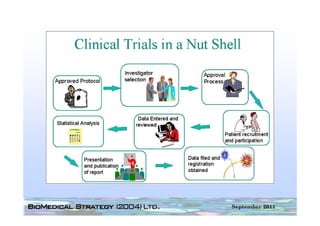






This document provides an overview of medical device regulation in the US and EU markets. It discusses key aspects of the regulatory process including intended use, classification, clinical evidence requirements, and approval pathways such as 510(k) and PMA. Major topics covered include regulatory strategies, claim determination, risk-based classification, clinical study design considerations, and substantial equivalence evaluations.


































































