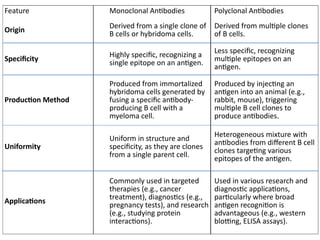
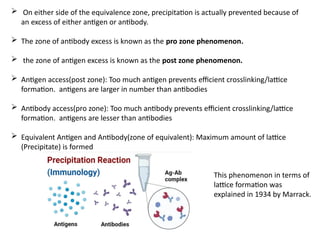
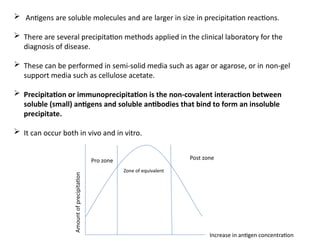
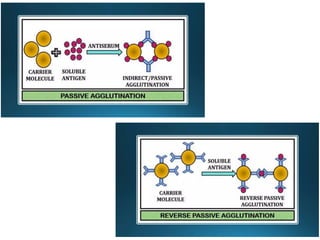
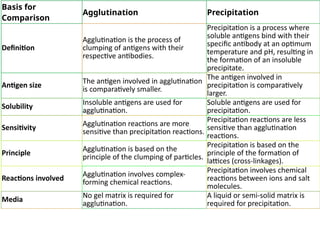
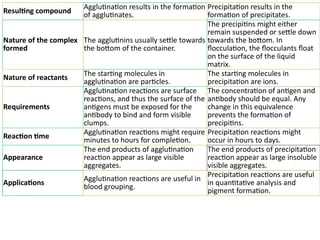
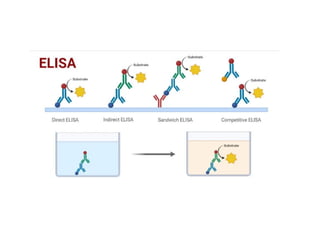
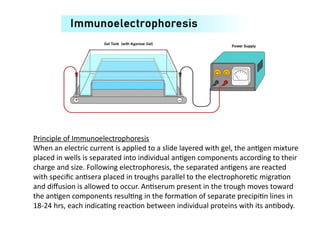
The document outlines serological techniques focusing on the interaction between antigens and antibodies, including definitions, types, and applications of immunoglobulins and the processes like agglutination and precipitation. It details various serological tests, particularly enzyme-linked immunosorbent assays (ELISA), which are instrumental in detecting immune responses through antigen-antibody interactions. Additionally, it compares methods of agglutination and precipitation, elaborating on the significance of these reactions in diagnostics and research.







































































































![谷歌留痕技术 [ 𝙩𝙤𝙥 𝟮𝟯𝟯. 𝙘 𝙤𝙢 ]](https://cdn.slidesharecdn.com/ss_thumbnails/top233-260130174328-3833018c-thumbnail.jpg?width=640&height=640&fit=bounds)



