Download as PDF, PPTX






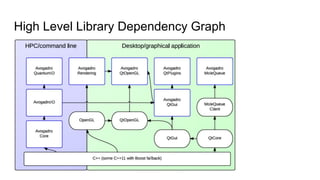





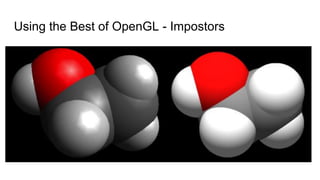




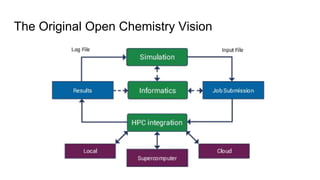



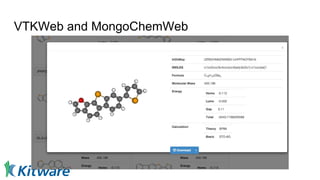

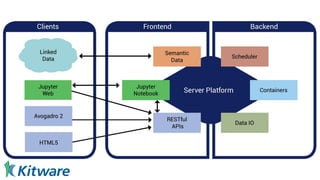

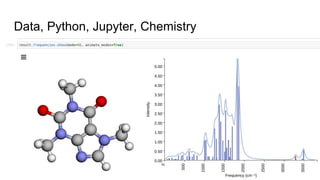


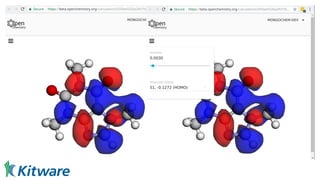
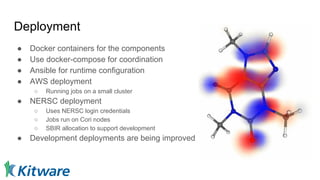

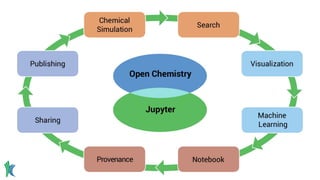





Dr. Marcus D. Hanwell discusses the development and history of Avogadro 2 and the Open Chemistry project, emphasizing its ambitious goals for enhancing molecular visualization and computational chemistry. He highlights the shift to a modular architecture, the integration of advanced rendering techniques, and the focus on extensibility and scalability. The presentation also covers the progress towards a more open platform that supports Jupyter integration and encourages community engagement in scientific research.










































![Polymer [ बहुलक ] Chemistry Notes PDF - Irfanullah Mehar - JJ Sir Chemistry.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/polymerchemistrynotespdf-irfanullahmehar-jjsirchemistry-260210172118-3f9b37f7-thumbnail.jpg?width=640&height=640&fit=bounds)


















