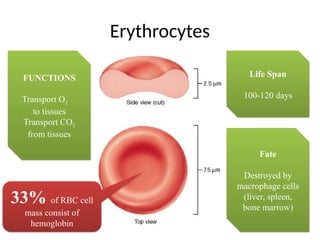
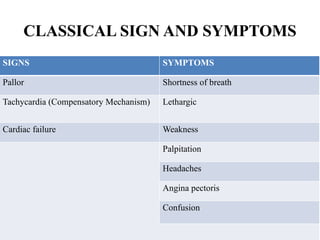
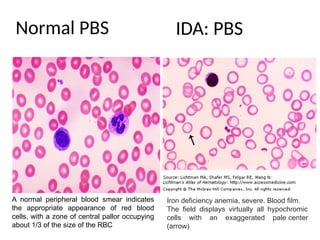
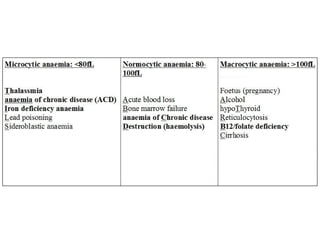
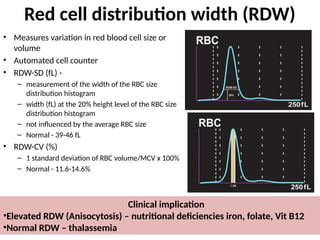
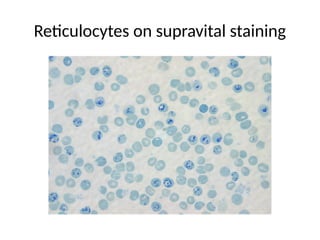
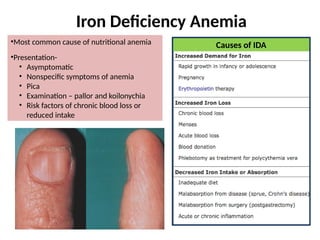
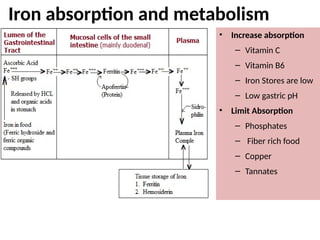
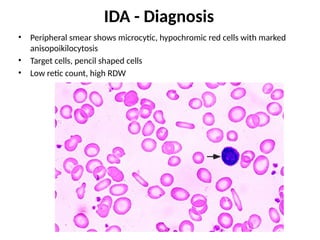
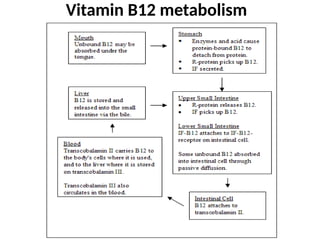
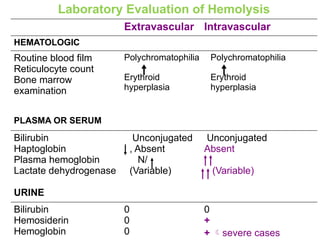
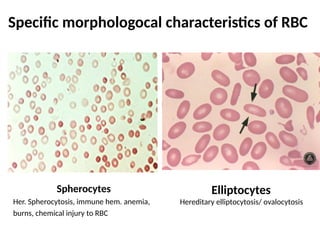
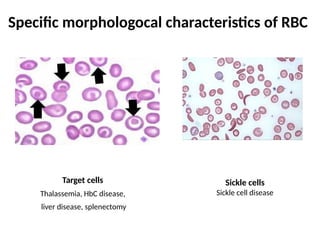
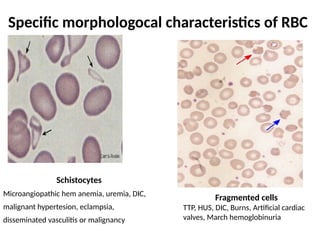
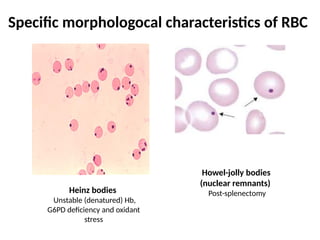
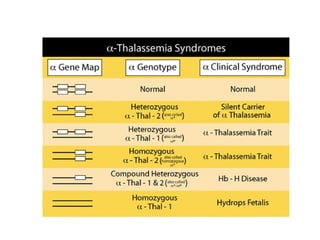
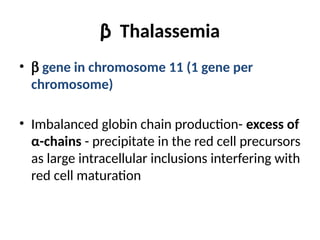
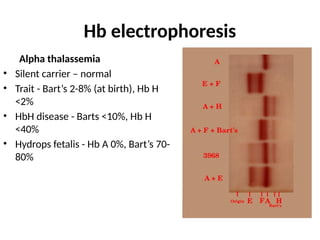
The document provides a comprehensive overview of anemia, including definitions, classifications, and causes of various types of anemia, as well as laboratory investigations and clinical presentations. It discusses specific types such as iron deficiency anemia, anemia of chronic disease, macrocytic anemias, and hemolytic anemias, alongside their respective pathophysiologies, diagnostic criteria, and treatment options. Key laboratory evaluations such as complete blood counts, peripheral blood smears, and reticulocyte counts are detailed to aid in clinical diagnosis and management of anemia.