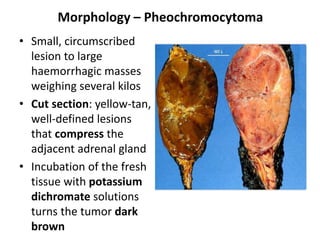
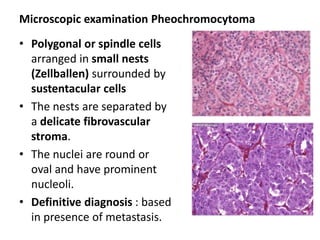
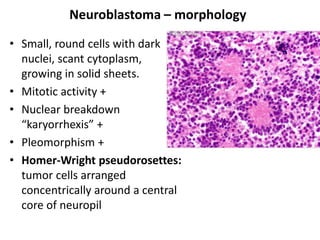
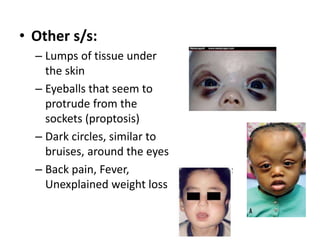
The document discusses adrenal medulla and tumors that can arise from it. It notes that adrenal medulla cells are derived from neural crest cells and their supporting cells. The two main tumors are neuroblastomas and pheochromocytomas. Pheochromocytomas arise from chromaffin cells and secrete catecholamines. They can be benign or malignant. Neuroblastomas are the most common extracranial solid tumor in childhood and commonly present with abdominal masses or chest symptoms. The document also discusses multiple endocrine neoplasia syndromes type 1 and 2.































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)