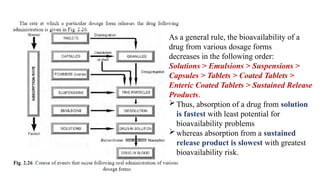
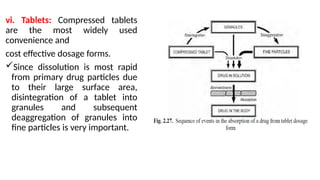
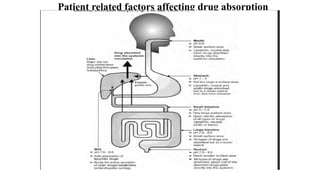
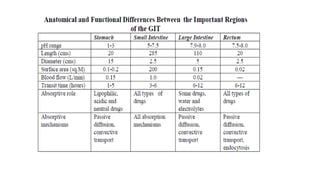
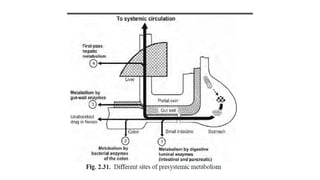
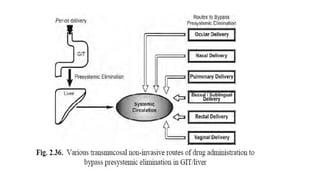
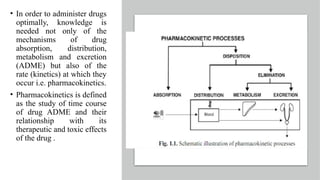
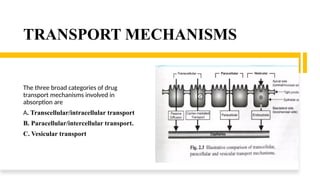
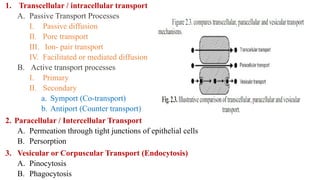
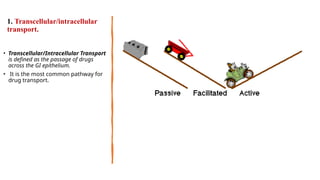
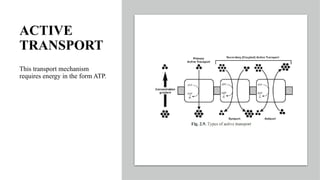
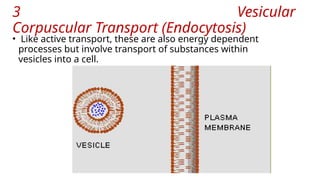
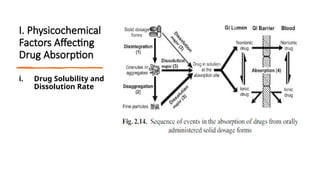
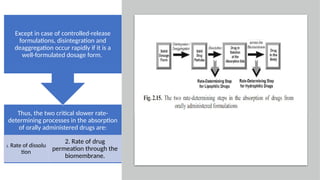
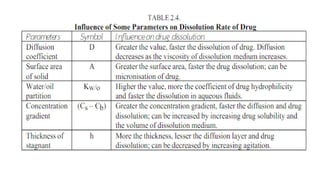
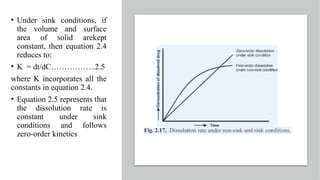
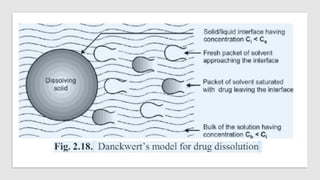
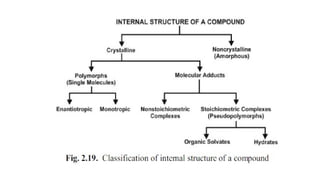
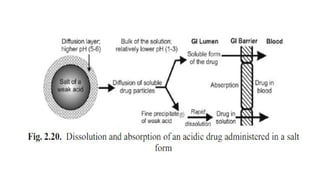
The document covers biopharmaceutics, focusing on drug absorption mechanisms and factors influencing drug bioavailability. It discusses various transport processes, including passive diffusion, facilitated diffusion, and active transport, as well as their relevance to drug formulation and administration. Additionally, it highlights the importance of pharmacokinetics in optimizing drug therapy and includes a detailed examination of factors affecting drug dissolution and absorption in the gastrointestinal tract.















































![2. Manufacturing/Processing Variables
The dosage form related factors that influence dissolution and hence absorption of a drug from such
formulations are:
1. Excipients (formulation ingredients apart from the active principles),
2. Manufacturing processes.
a. Method of granulation, and
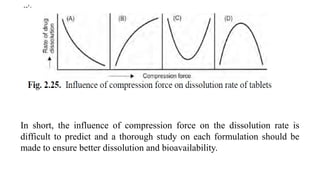
b. Compression force- The compression force employed in tabletting process influence
density, porosity, hardness, disintegration time and dissolution of tablets.
On the one hand, higher compression force increases the density and hardness of tablet,
decreases porosity and hence penetrability of the solvent into the tablet, retards wettability by
forming a firmer and more effective sealing layer by the lubricant, and in many cases,
promotes tighter bonding between the particles, all of which result in slowing of the
dissolution rate of tablets.[curve A]
On the other hand, higher compression forces cause deformation, crushing or fracture of drug
particles into smaller ones or convert a spherical granule into a disc shaped particle with a
large increase in the effective surface area. This results in an increase in the dissolution rate of
the tablet.[curve B]
A combination of both the curves A and B is also possible as shown in curves C and D.](https://image.slidesharecdn.com/ch1bip-250128103038-9e41d337/85/Absorption-Biopharmaceutics-and-Pharmacokinetics-60-320.jpg)