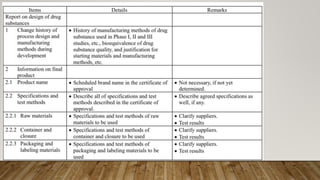
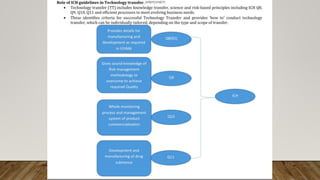
This document discusses technology transfer from R&D to production. It defines technology transfer and explains its importance for moving from drug discovery to commercialization. It outlines various stages of formulation development and discusses procedures for technology transfer including development reports, product specifications, technology transfer plans and documentation. It emphasizes the importance of communication between transferring and receiving parties and quality assurance approval. Technical information to be transferred includes details on facilities, equipment, manufacturing methods and test methods.