Download as PDF, PPTX

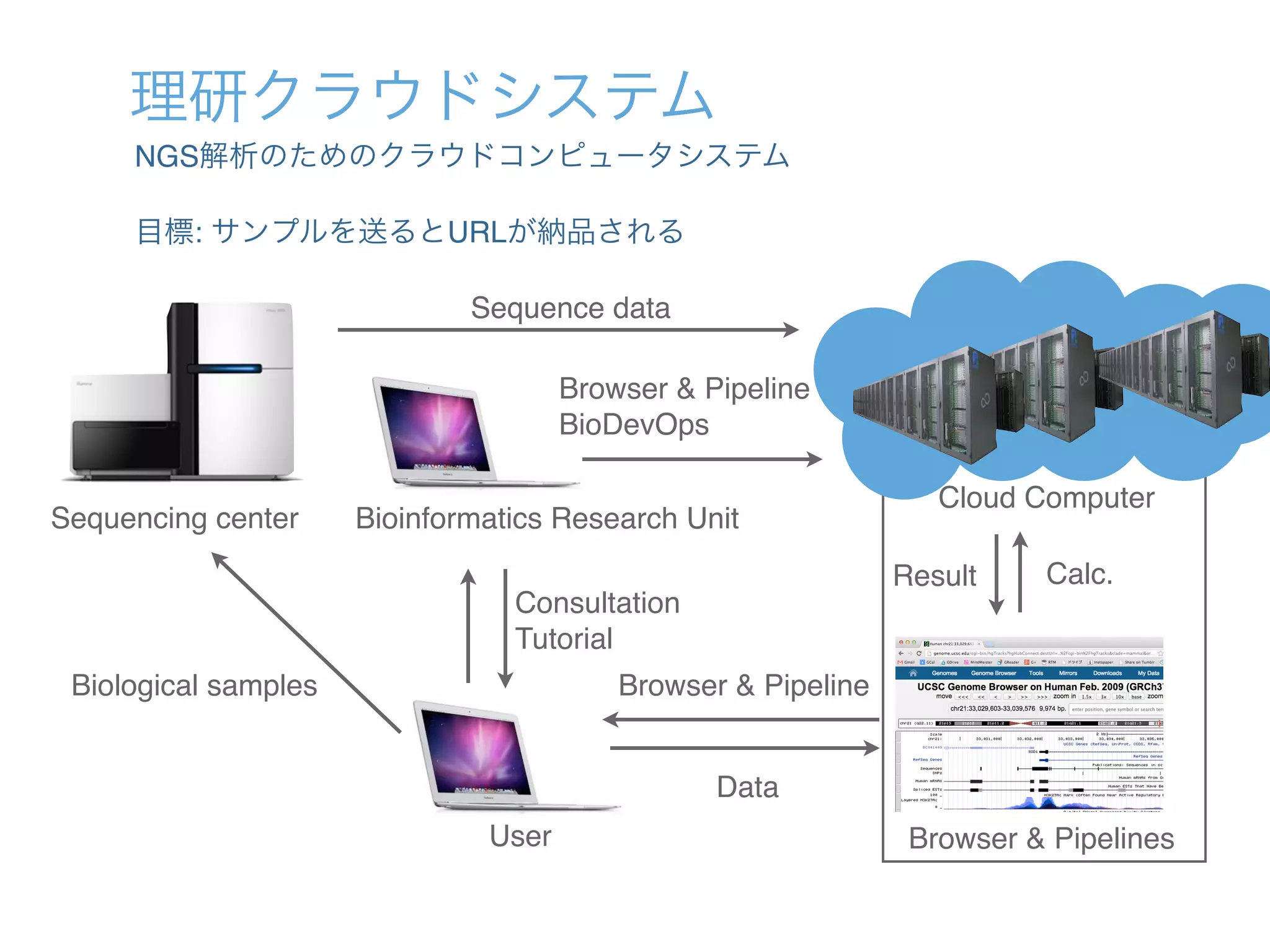
![国立遺伝学研究所スーパーコンピュータよる成果
Thanks!
METHOD Open Access
Quartz-Seq: a highly reproducible and sensitive
single-cell RNA sequencing method, reveals non-
genetic gene-expression heterogeneity
Yohei Sasagawa1,7†
, Itoshi Nikaido1,7†
, Tetsutaro Hayashi2
, Hiroki Danno3
, Kenichiro D Uno1
, Takeshi Imai4,5
and
Hiroki R Ueda1,3,6*
Abstract
Development of a highly reproducible and sensitive single-cell RNA sequencing (RNA-seq) method would facilitate
the understanding of the biological roles and underlying mechanisms of non-genetic cellular heterogeneity. In this
study, we report a novel single-cell RNA-seq method called Quartz-Seq that has a simpler protocol and higher
reproducibility and sensitivity than existing methods. We show that single-cell Quartz-Seq can quantitatively detect
various kinds of non-genetic cellular heterogeneity, and can detect different cell types and different cell-cycle
phases of a single cell type. Moreover, this method can comprehensively reveal gene-expression heterogeneity
between single cells of the same cell type in the same cell-cycle phase.
Keywords: Single cell, RNA-seq, Transcriptome, Sequencing, Bioinformatics, Cellular heterogeneity, Cell biology
Background
Non-genetic cellular heterogeneity at the mRNA and pro-
tein levels has been observed within cell populations in
diverse developmental processes and physiological condi-
tions [1-4]. However, the comprehensive and quantitative
analysis of this cellular heterogeneity and its changes in
response to perturbations has been extremely challenging.
Recently, several researchers reported quantification of
gene-expression heterogeneity within genetically identical
cell populations, and elucidation of its biological roles and
underlying mechanisms [5-8]. Although gene-expression
heterogeneities have been quantitatively measured for sev-
eral target genes using single-molecule imaging or single-
cell quantitative (q)PCR, comprehensive studies on the
quantification of gene-expression heterogeneity are limited
[9] and thus further work is required. Because global
gene-expression heterogeneity may provide biological
information (for example, on cell fate, culture environ-
ment, and drug response), the question of how to compre-
hensively and quantitatively detect the heterogeneity of
mRNA expression in single cells and how to extract biolo-
gical information from those data remains to be addressed.
Single-cell RNA sequencing (RNA-seq) analysis has
been shown to be an effective approach for the compre-
hensive quantification of gene-expression heterogeneity
that reflects the cellular heterogeneity at the single-cell
level [10,11]. To understand the biological roles and
underlying mechanisms of such heterogeneity, an ideal
single-cell transcriptome analysis method would provide
a simple, highly reproducible, and sensitive method for
measuring the gene-expression heterogeneity of cell
populations. In addition, this method should be able to
distinguish clearly the gene-expression heterogeneity
from experimental errors.
Single-cell transcriptome analyses, which can be
achieved through the use of various platforms, such as
microarrays, massively parallel sequencers and bead arrays
[12-17], are able to identify cell-type markers and/or rare
cell types in tissues. These platforms require nanogram
quantities of DNA as the starting material. However, a
typical single cell has approximately 10 pg of total RNA
and often contains only 0.1 pg of polyadenylated RNA,
hence, o obtain the amount of DNA starting material that
is required by these platforms, it is necessary to perform
whole-transcript amplification (WTA).
* Correspondence: uedah-tky@umin.ac.jp
† Contributed equally
1
Functional Genomics Unit, RIKEN Center for Developmental Biology, 2-2-3
Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan
Full list of author information is available at the end of the article
Sasagawa et al. Genome Biology 2013, 14:R31
http://genomebiology.com/2013/14/4/R31
© 2013 Sasagawa et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative
Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Molecular Cell
Article
Context-Dependent Wiring of Sox2 Regulatory
Networks for Self-Renewal
of Embryonic and Trophoblast Stem Cells
Kenjiro Adachi,1,9,10,* Itoshi Nikaido,2,9,11 Hiroshi Ohta,3,12 Satoshi Ohtsuka,1 Hiroki Ura,1 Mitsutaka Kadota,5
Teruhiko Wakayama,3,6 Hiroki R. Ueda,2,4 and Hitoshi Niwa1,7,8,*
1Laboratory for Pluripotent Stem Cell Studies
2Functional Genomics Unit
3Laboratory for Genome Reprogramming
4Laboratory for Systems Biology
5Genome Resource and Analysis Unit
RIKEN Center for Developmental Biology, 2-2-3 Minatojima-minamimachi, Chuo-ku, Kobe 6500047, Japan
6Faculty of Life and Environmental Sciences, University of Yamanashi, Yamanashi 4008510, Japan
7Laboratory for Development and Regenerative Medicine, Kobe University Graduate School of Medicine, 7-5-1 Kusunokicho, Chuo-ku,
Kobe 6500017, Japan
8JST, CREST, Sanbancho, Chiyoda-ku, Tokyo, 1020075, Japan
9These authors contributed equally to this work
10Present address: Department of Cell and Developmental Biology, Max Planck Institute for Molecular Biomedicine, 48149 Mu¨ nster, Germany
11Present address: Bioinformatics Research Unit, Advanced Center for Computing and Communication, RIKEN, Wako,
Saitama 3510198, Japan
12Present address: Department of Anatomy and Cell Biology, Graduate School of Medicine, Kyoto University, Sakyo-ku,
Kyoto 6068501, Japan
*Correspondence: kenjiro.adachi@mpi-muenster.mpg.de (K.A.), niwa@cdb.riken.jp (H.N.)
http://dx.doi.org/10.1016/j.molcel.2013.09.002
SUMMARY
Sox2 is a transcription factor required for the mainte-
nance of pluripotency. It also plays an essential role
in different types of multipotent stem cells, raising
the possibility that Sox2 governs the common
stemness phenotype. Here we show that Sox2 is
a critical downstream target of fibroblast growth fac-
tor (FGF) signaling, which mediates self-renewal of
trophoblast stem cells (TSCs). Sustained expression
of Sox2 together with Esrrb or Tfap2c can replace
FGF dependency. By comparing genome-wide bind-
ing sites of Sox2 in embryonic stem cells (ESCs) and
TSCs combined with inducible knockout systems,
we found that, despite the common role in safe-
guarding the stem cell state, Sox2 regulates distinct
sets of genes with unique functions in these two
different yet developmentally related types of
stem cells. Our findings provide insights into the
functional versatility of transcription factors during
embryogenesis, during which they can be recur-
sively utilized in a variable manner within discrete
network structures.
INTRODUCTION
The transcriptional output of a given cell type is controlled by
unique combinations of transcription factors under the control
of extrinsic signals that can modulate the expression and activity
of transcription factors, forming a gene regulatory network that
dictates a specific cellular phenotype. Tissue-specific transcrip-
tion factors play deterministic roles in cell-type specification,
which is manifested as lineage reprogramming by forced expres-
sion of such transcription factors (Graf and Enver, 2009; Zhou
and Melton, 2008). Sox2 is one such transcription factor required
for the maintenance of pluripotent stem cells in vivo (Avilion et al.,
2003) and in vitro (Masui et al., 2007) and for the induction of
pluripotency (Takahashi and Yamanaka, 2006). However, it is
also preferentially expressed in neural, retinal, and trophoblast
stem cells (TSCs) (Avilion et al., 2003; Pevny and Nicolis,
2010), suggesting a possible role for Sox2 in governing a com-
mon stemness phenotype.
In embryonic stem cells (ESCs), Sox2 forms a heterodimer
with Oct3/4 (also known as Pou5f1) on DNA with the OCT-SOX
composite motifs, and these factors cooperatively activate
pluripotency-related target genes such as Nanog, Fgf4, Utf1,
Lefty1, and Fbxo15, as well as their own expression (Nakatake
et al., 2006, and references therein). Oct3/4-knockout ESCs
are differentiated along the trophoblast lineage in a highly homo-
geneous manner (Niwa et al., 2000). In contrast, the loss of Sox2
causes differentiation of ESCs accompanied by upregulation of
markers for trophoblast and embryonic germ layers, although
artificial maintenance of Oct3/4 from the transgene can sustain
self-renewal and pluripotency of Sox2-null ESCs (Masui et al.,
2007), suggesting that the unique function of Sox2 may be
to maintain Oct3/4 expression. These two core transcription
factors, along with Nanog, form an interconnected and hierarchi-
cal network downstream of the leukemia inhibitory factor (LIF)-
Stat3 and LIF-phosphatidylinositol 3-kinase (PI3K) signaling
380 Molecular Cell 52, 380–392, November 7, 2013 ª2013 Elsevier Inc.
世界最高精度の1細胞RNA-Seq開発 転写因子ネットワークの動的変化と分化](https://image.slidesharecdn.com/20140731nigscuserspublic-140807024650-phpapp02/75/DevOps-cloud-DNA-2-2048.jpg)
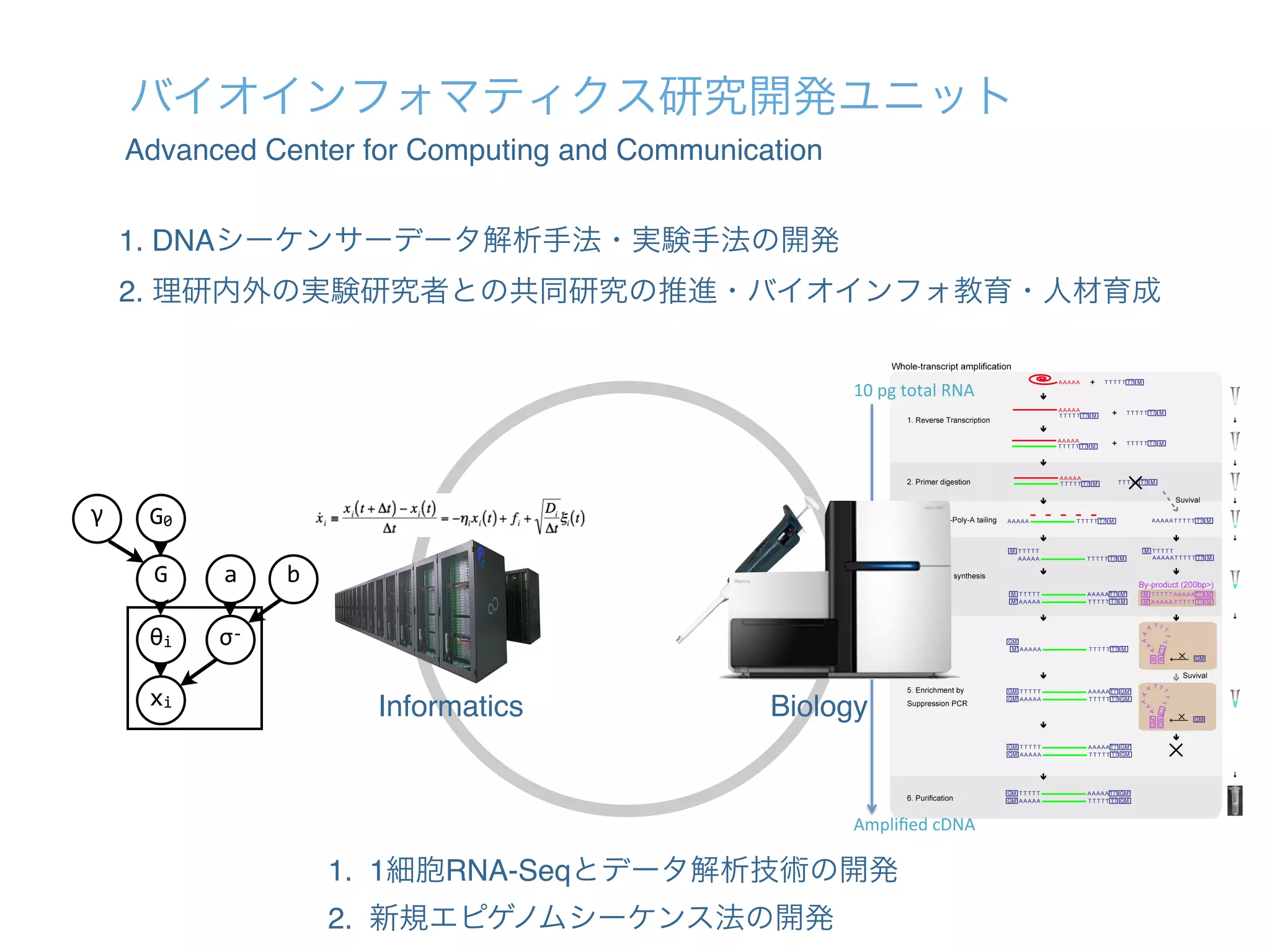

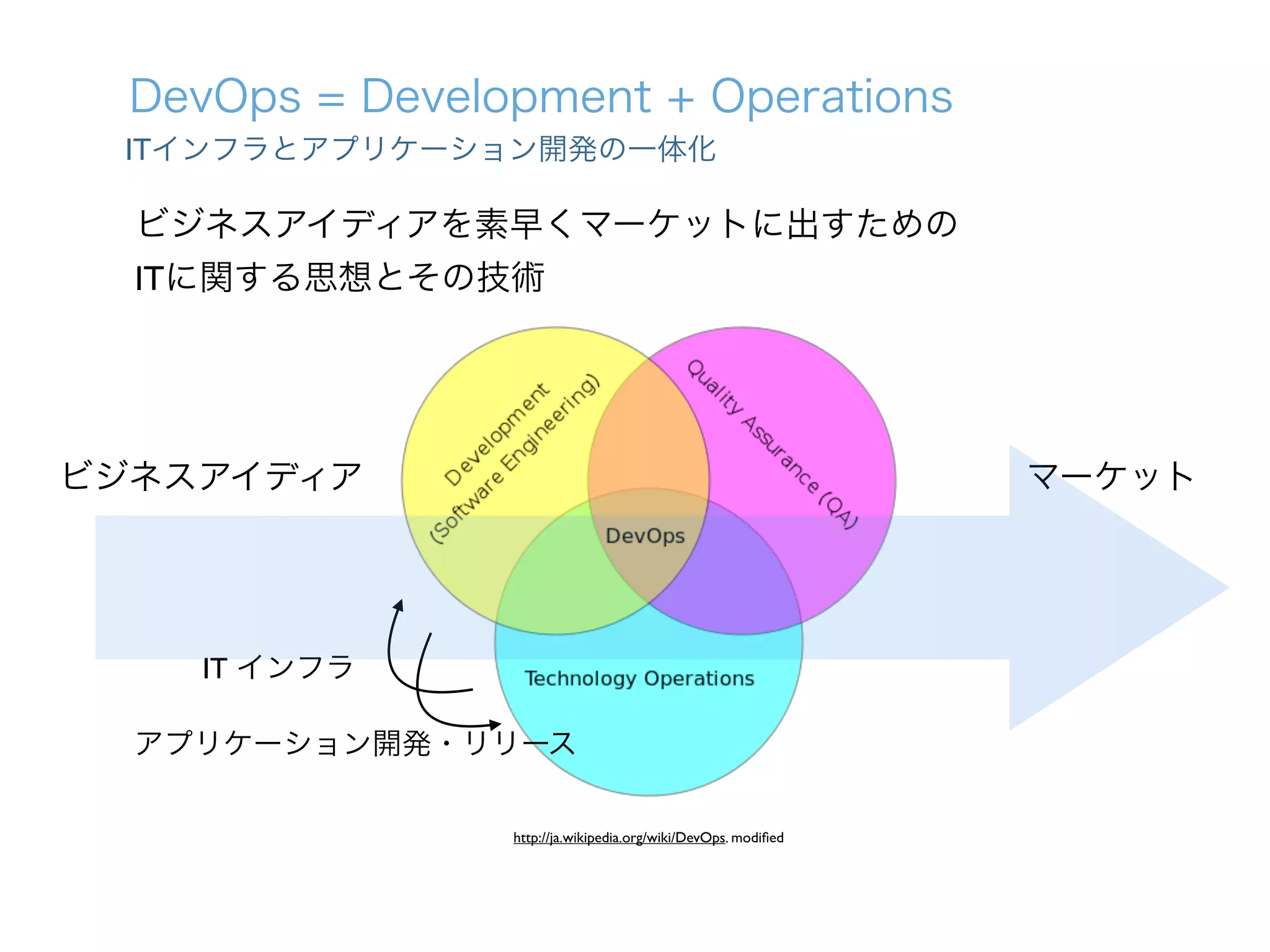
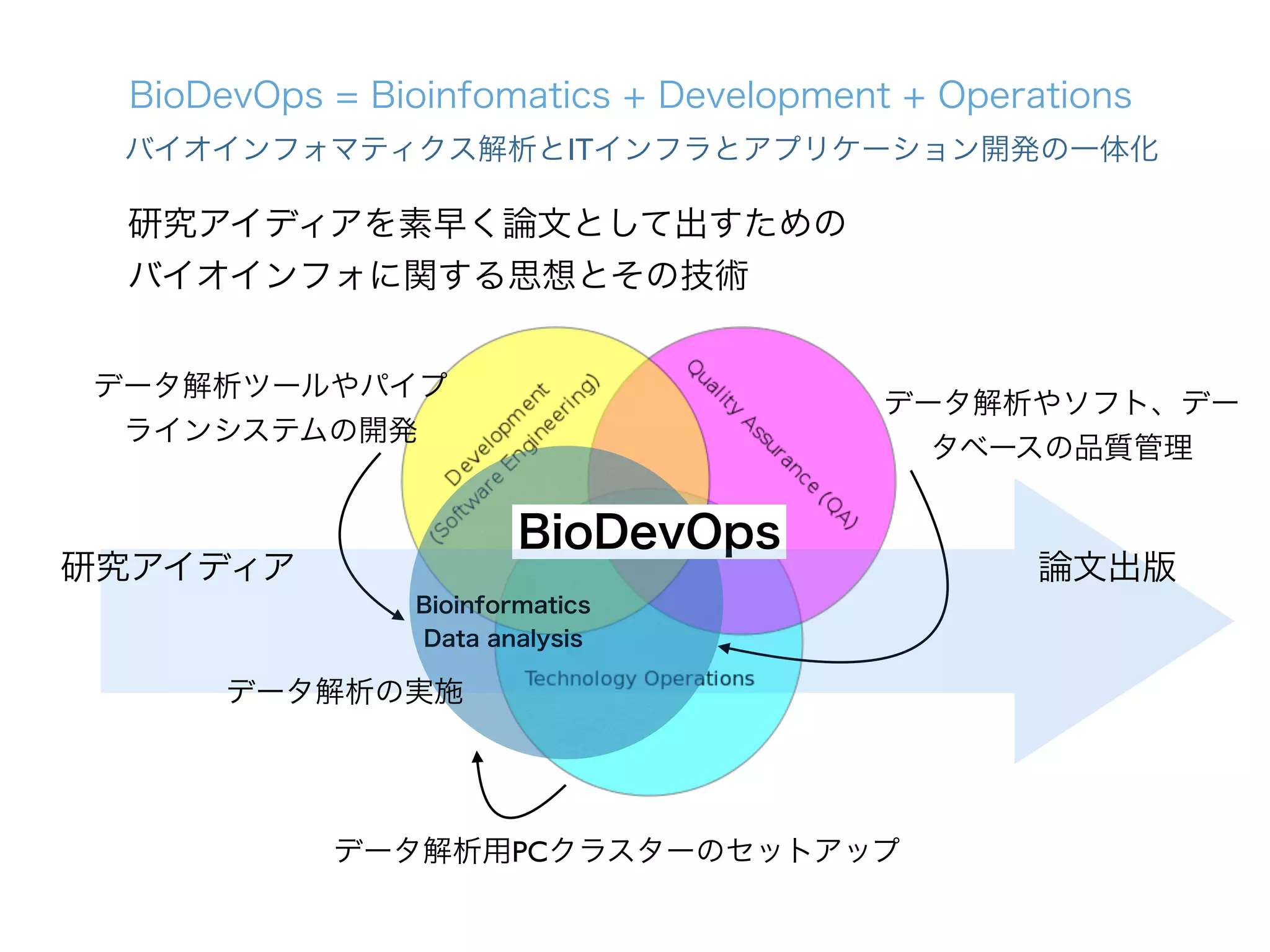

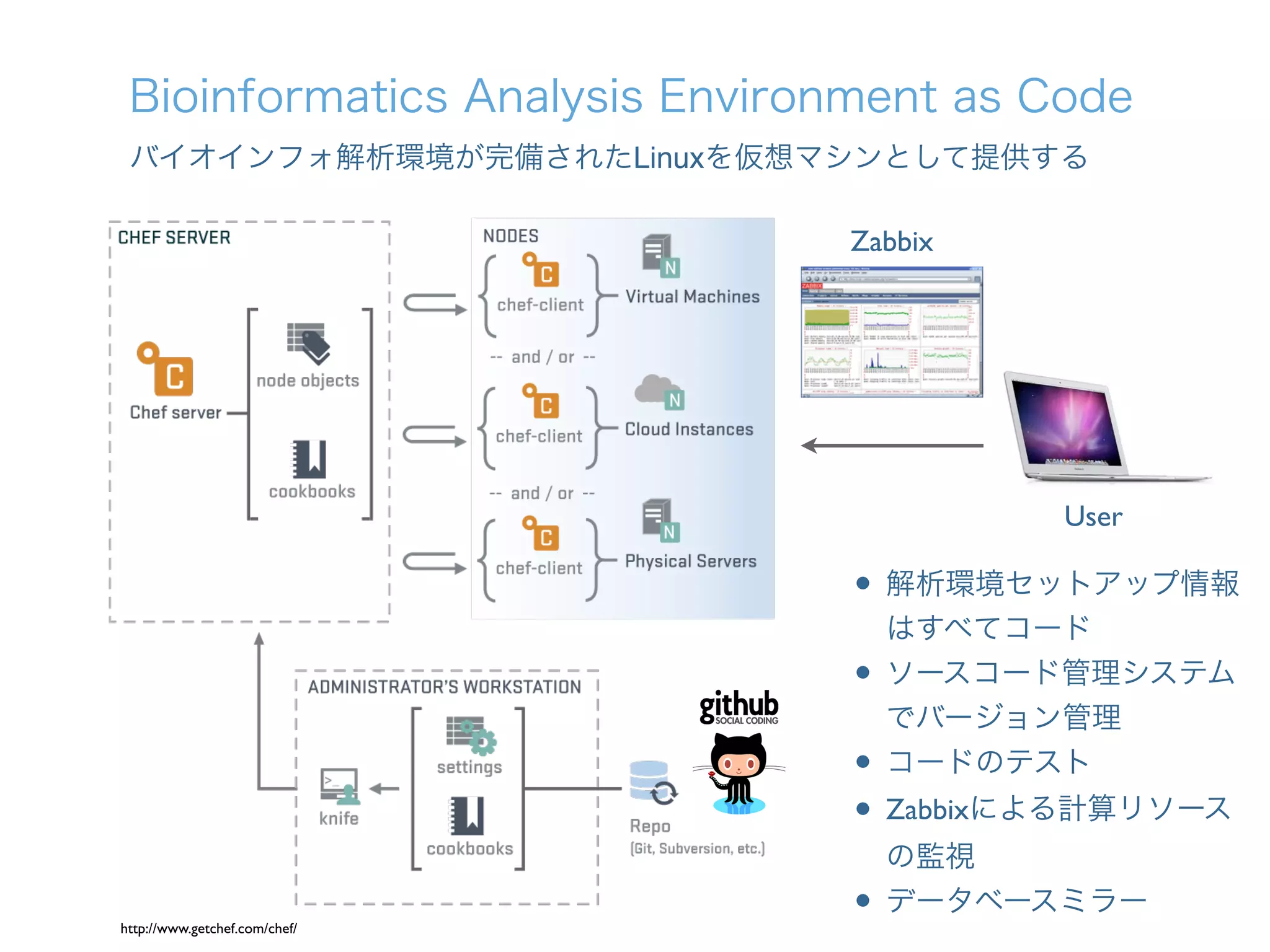



1. The document describes the development of a new single-cell RNA-seq method called Quartz-Seq that has higher reproducibility and sensitivity than existing methods. 2. Quartz-Seq can quantitatively detect various types of non-genetic cellular heterogeneity and can distinguish different cell types and cell cycle phases of a single cell type. 3. It can also comprehensively reveal gene expression heterogeneity between single cells of the same cell type in the same cell cycle phase.




























































![[DSC Europe 25] Tatevik Maytesyan - How to actually use AI in marketing: gett...](https://cdn.slidesharecdn.com/ss_thumbnails/tjo626lsqdgfntbgl2mw-4-251216103155-e36cd239-thumbnail.jpg?width=640&height=640&fit=bounds)




![[DSC Europe 25] Uros Pesic - The Reality of AI in Marketing.pdf](https://cdn.slidesharecdn.com/ss_thumbnails/rtkodnmtycovsllvzsyn-9-251215095918-b0c6bfe3-thumbnail.jpg?width=640&height=640&fit=bounds)






![[DSC Europe 25] Branko Dzakula - From Defense to Attack: How AI Redefines Cyb...](https://cdn.slidesharecdn.com/ss_thumbnails/80bdzdxpr3ky2g0qvyk9-8-251211083048-ce5fc1ee-thumbnail.jpg?width=640&height=640&fit=bounds)

![[DSC Europe 25] Behzad Hosseini - AI Agents in the Wild: Deploying Models tha...](https://cdn.slidesharecdn.com/ss_thumbnails/3qtejajvsjqrzwfept2c-10-251212103250-7f2b1068-thumbnail.jpg?width=640&height=640&fit=bounds)
![[DSC Europe 25] Jovan Bogicevic - Legacy to AI-Driven Defense: Transforming D...](https://cdn.slidesharecdn.com/ss_thumbnails/rsarluadt563hntyfc8q-3-251211083849-3e7bc4c0-thumbnail.jpg?width=640&height=640&fit=bounds)
