
Download as PDF, PPTX





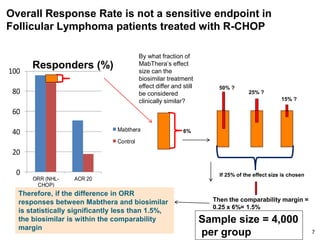
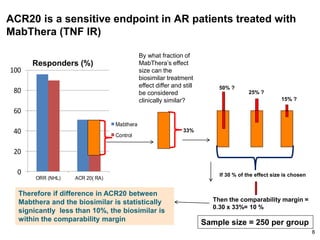

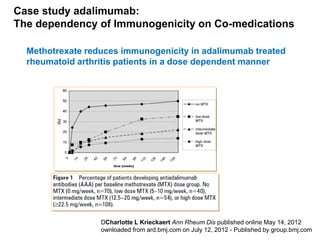

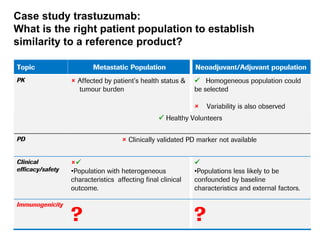
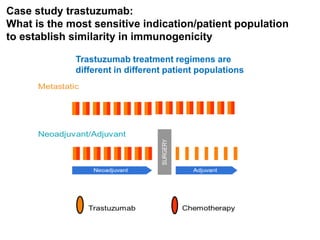
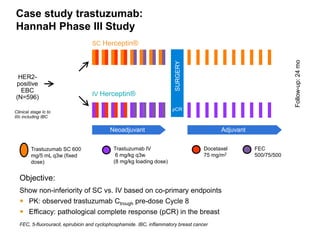



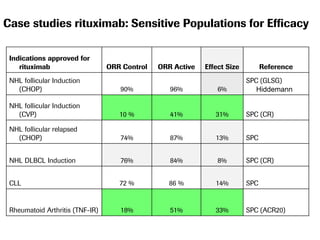

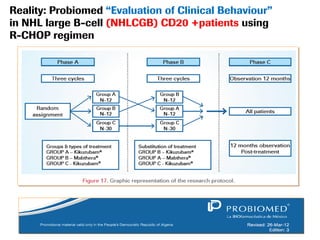


The document discusses the importance of selecting sensitive patient populations for clinical trials of biosimilar monoclonal antibodies to properly assess clinical similarity to the reference product. It provides examples of indications and populations that are more sensitive for detecting differences based on effect size, such as rheumatoid arthritis patients for assessing ACR20 response to anti-TNF antibodies. The document cautions that clinical trials must be designed appropriately to demonstrate equivalence or non-inferiority and should obtain data on relevant endpoints in sensitive populations as well as long-term safety and immunogenicity follow-up to justify extrapolation to other indications. It critiques examples of biosimilar clinical trials that failed to use a sensitive population or design.








































































































