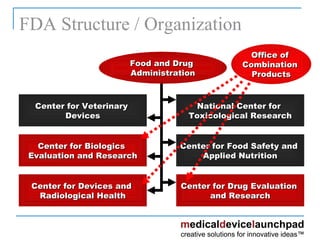
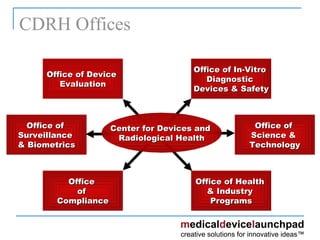
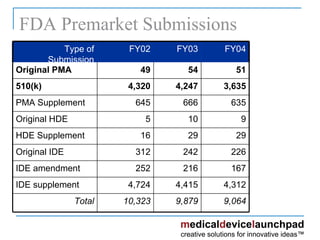
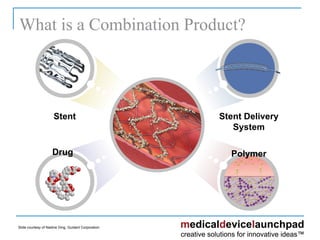
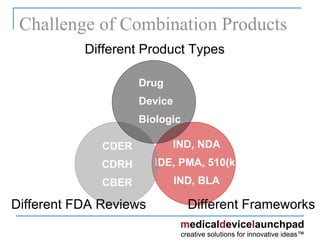
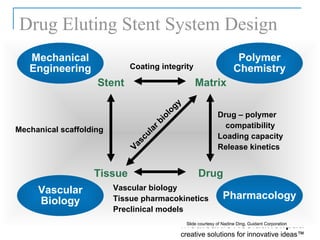
The document provides an overview of the FDA's regulatory framework for medical devices, outlining device classifications, submission types, and approval requirements. It details the 510(k) and PMA processes for device marketing authorization and the roles of different FDA centers in managing these products. Additionally, it discusses investigational device exemptions and challenges associated with combination products encompassing drugs and biologics.