Downloaded 10 times









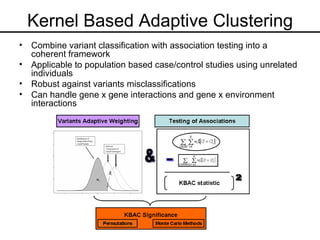

This document discusses statistical genetics and the use of statistics in analyzing genetic sequence data. It covers several key areas: the origins of statistical genetics in the work of Fisher, Galton and Pearson; methods for disease gene mapping including linkage analysis and genome-wide association studies; challenges in analyzing sequencing data including rare variant detection and accounting for interactions; and hypotheses for multi-factorial disease etiology including the common disease-common variant and common disease-rare variant hypotheses. It also describes challenges in statistical methodologies for variant classification and handling interactions and proposes a kernel-based adaptive clustering approach.






















































































