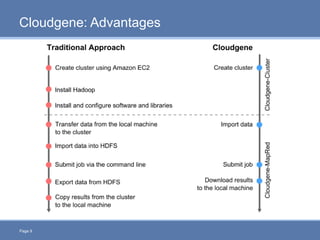
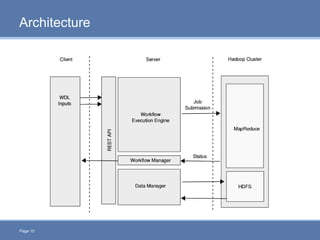
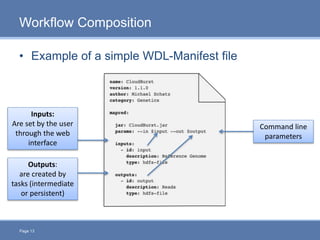
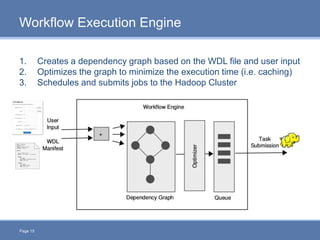
CloudGene is a graphical workflow management system designed for MapReduce applications in bioinformatics, enabling non-experts to create and execute workflows for analyzing large genomic datasets. It integrates existing MapReduce tools without the need for source code adaptations and operates in a web-based environment, requiring a compatible cluster or leveraging cloud computing. The system supports a variety of technologies including Apache Hadoop, Apache Pig, and offers features such as user management and parameter tracking.






































![Resource Aware Scheduling for Hadoop [Final Presentation]](https://cdn.slidesharecdn.com/ss_thumbnails/fyppresentationfinal-120226092754-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)




















































