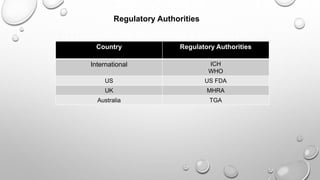
This document discusses quality assurance and quality control in the pharmaceutical industry. It defines quality assurance and quality control, explaining that QA involves planned review procedures while QC involves routine technical activities to measure and control quality. It discusses Good Laboratory Practice (GLP), Good Manufacturing Practice (GMP), and ICH guidelines. It provides details on in-process quality control tests for various drug formulations and finished product testing. It also discusses regulatory authorities, documentation, instrumentation handling, validation, calibration, and qualification which are important aspects of QA and QC in the pharmaceutical industry.