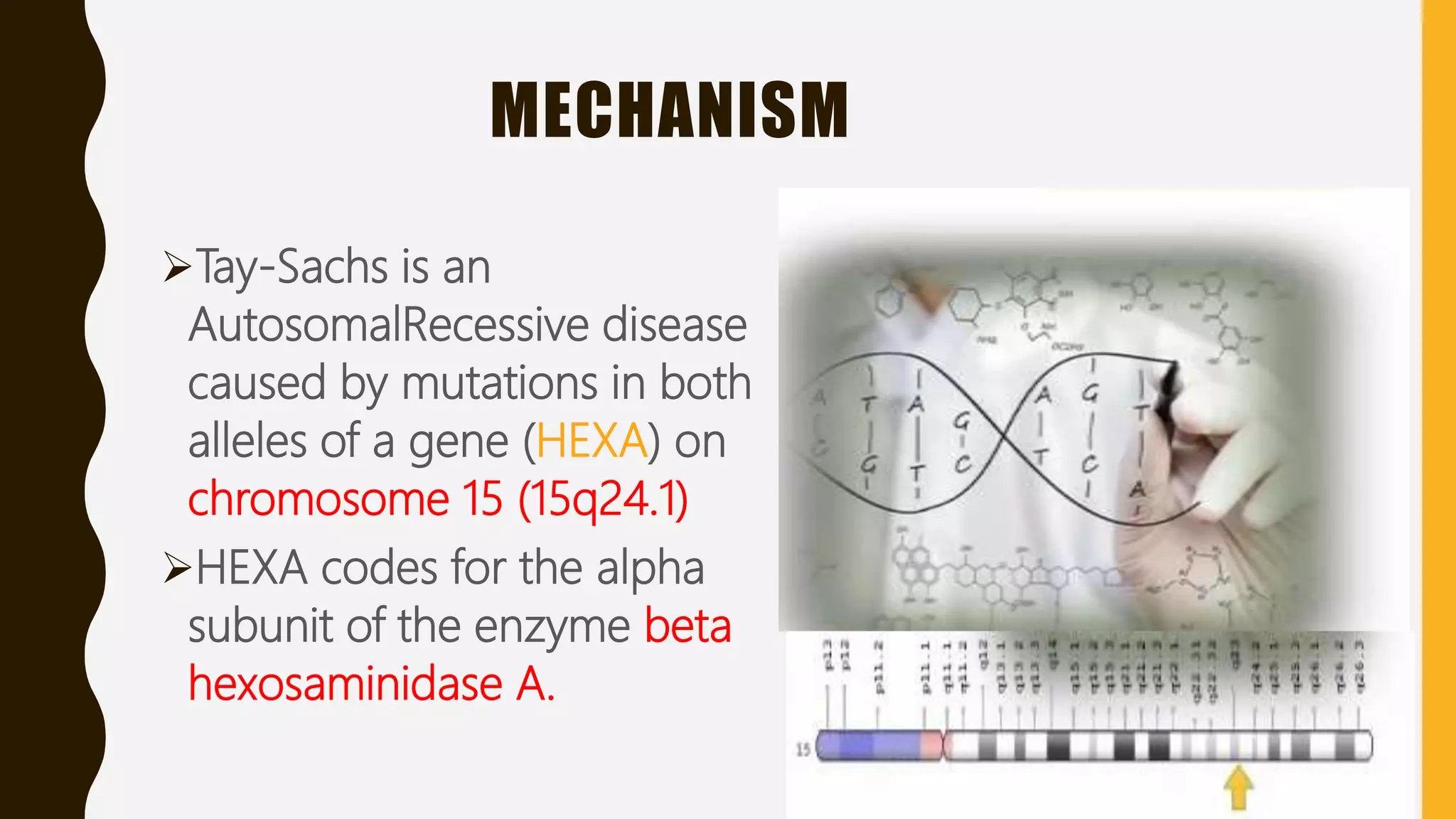
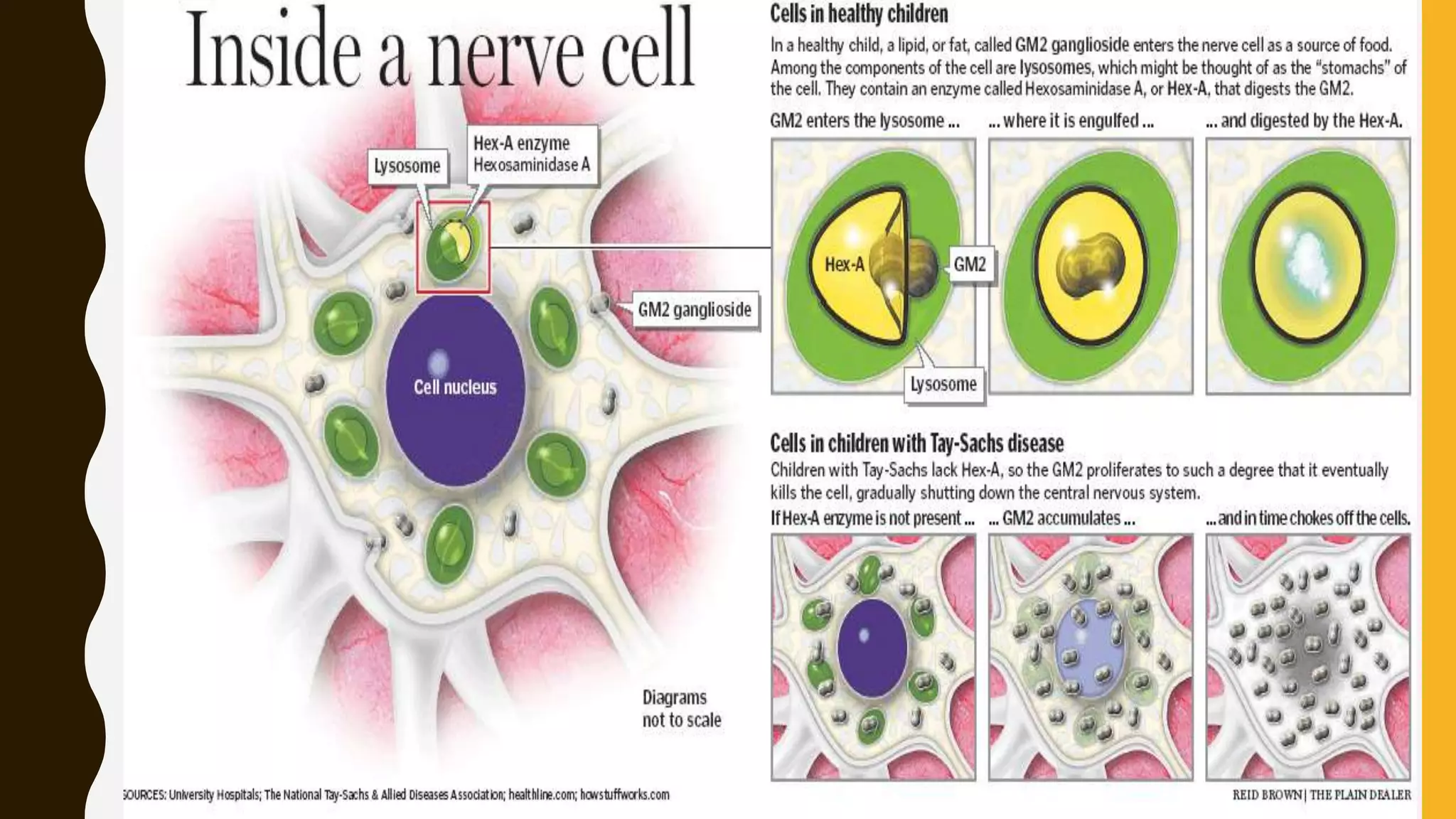
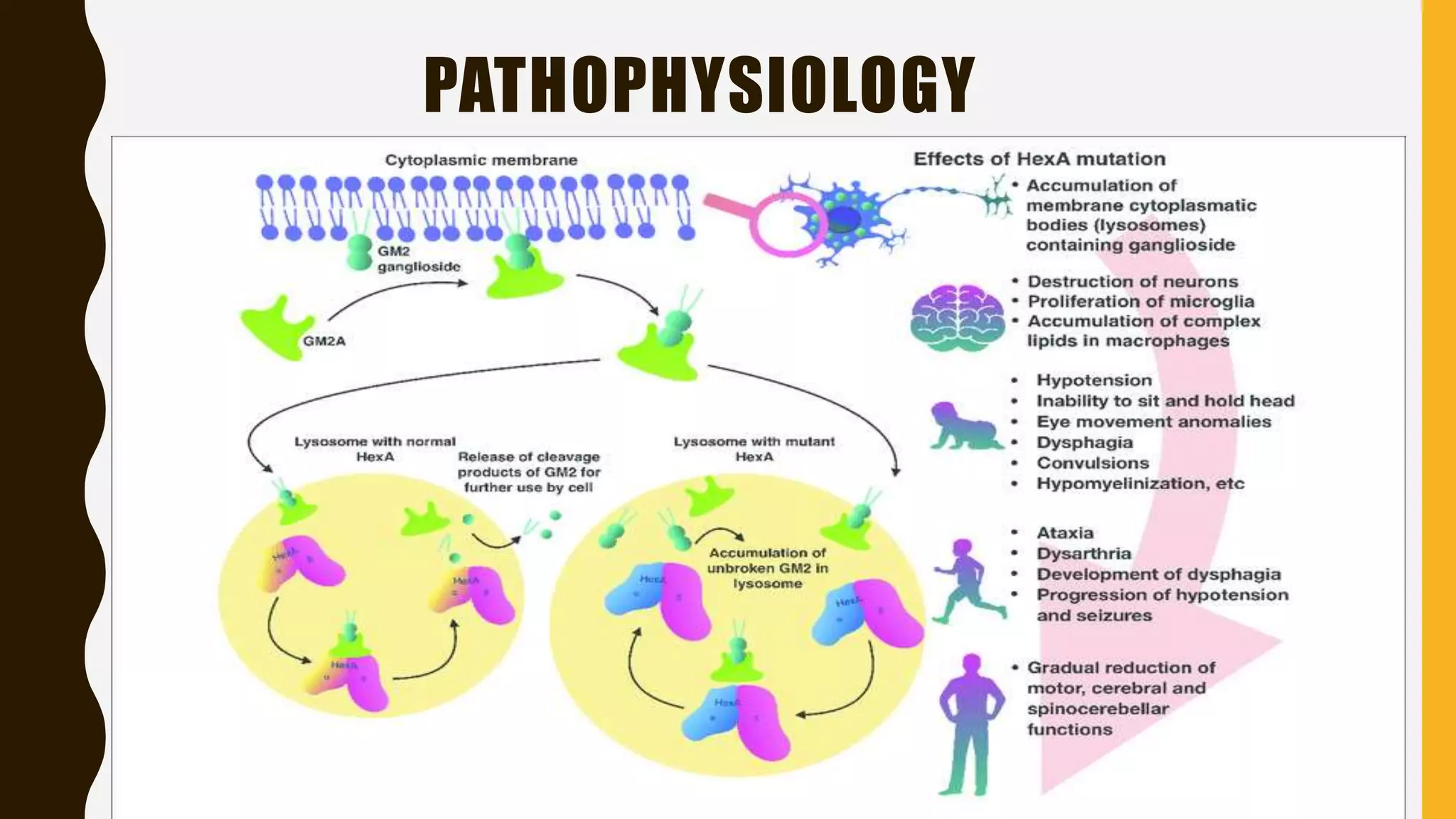
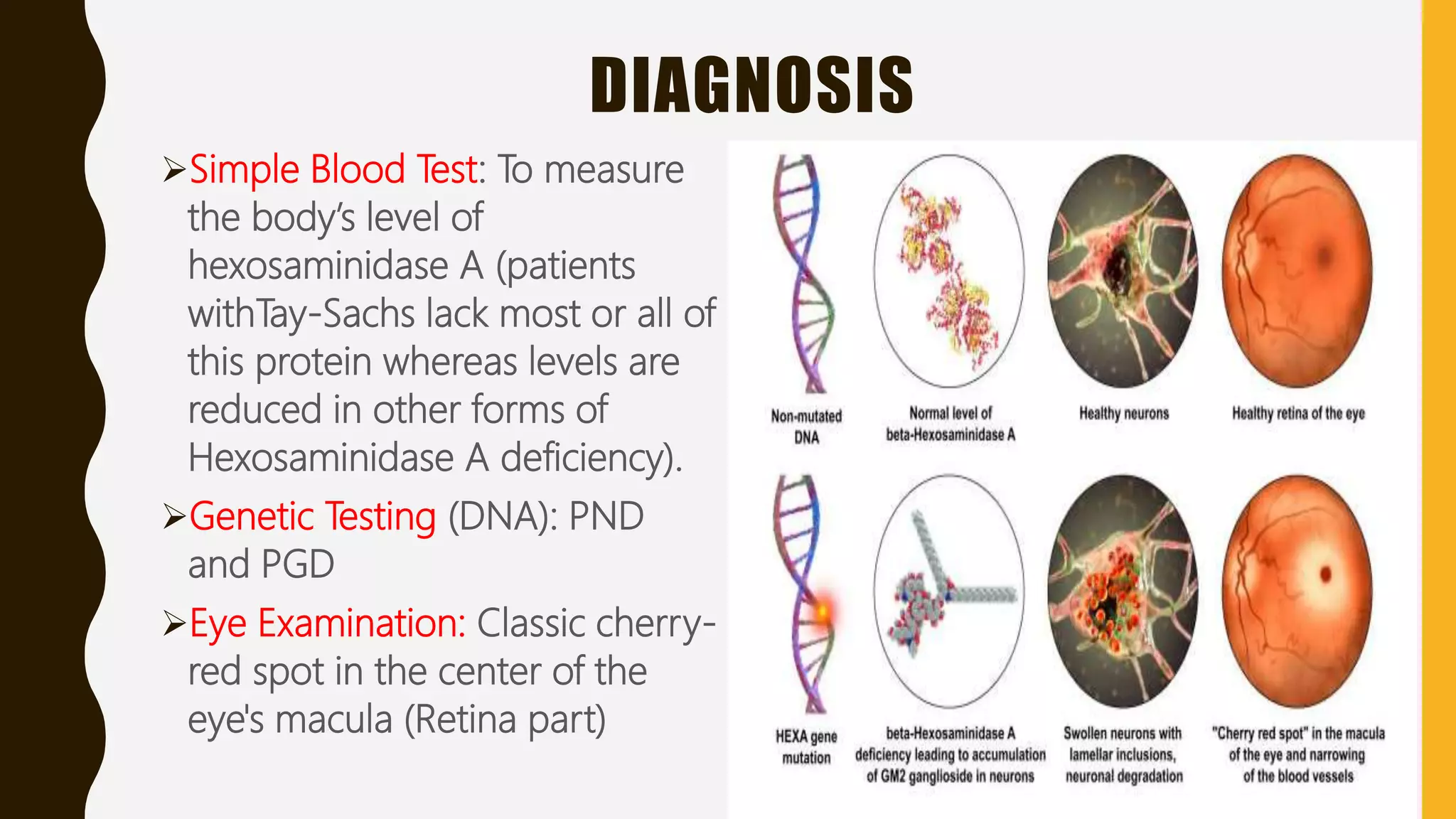
Tay-Sachs disease is a rare autosomal recessive disorder caused by mutations in the HEXA gene on chromosome 15, leading to a deficiency in the enzyme beta-hexosaminidase A, resulting in toxic levels of GM2 ganglioside that affect nerve cell function. The disease is classified into three types based on symptom onset: infantile, juvenile, and adult/late onset, with varying life expectancies. Diagnosis involves blood tests for enzyme levels, genetic testing, and eye examinations, while treatment focuses on supportive care, as there is currently no cure.