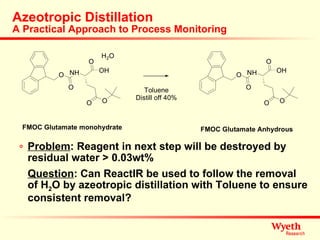
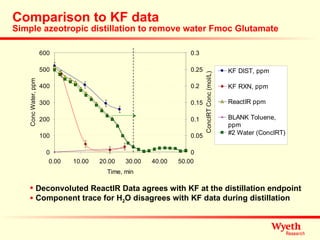
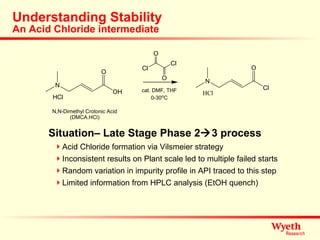
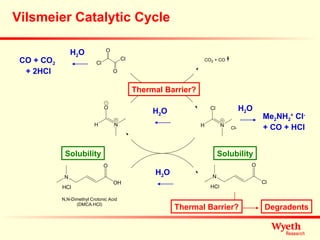
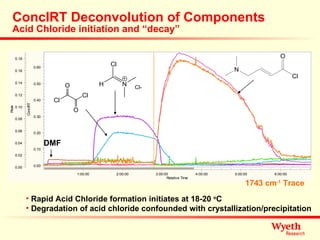
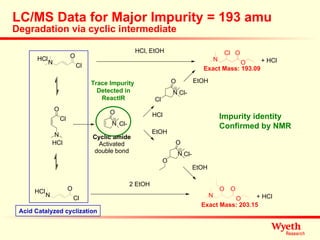
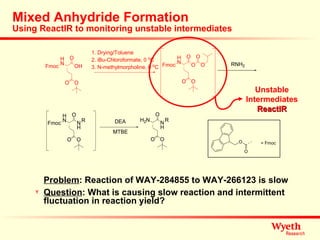
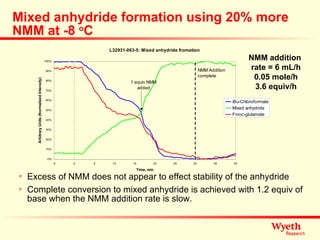
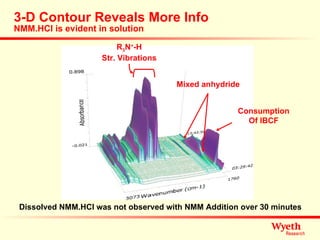
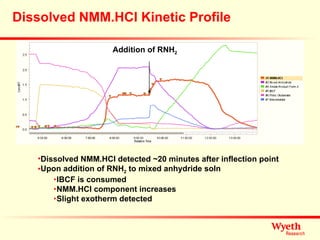
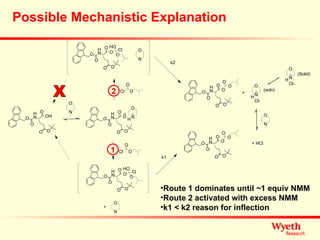
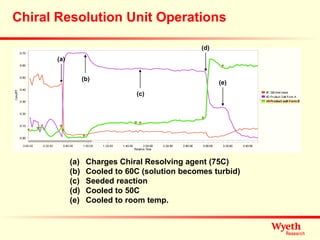
The document discusses using ReactIR technology to provide insights into chemical reactions and processes. It presents three case studies where ReactIR was used: (1) monitoring an unstable acid chloride intermediate in a Vilsmeier reaction, (2) studying mixed anhydride formation with unstable intermediates, and (3) gaining understanding of a chiral resolution process. ReactIR allowed observing reaction components in real-time, identifying side reactions, and gaining mechanistic insights in all three cases.