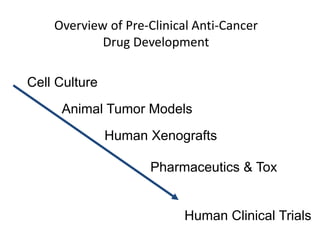
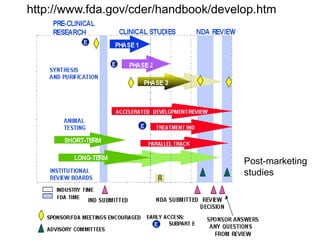
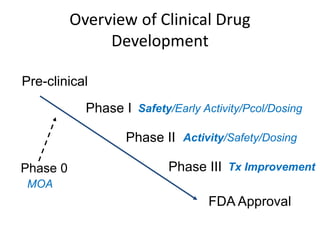
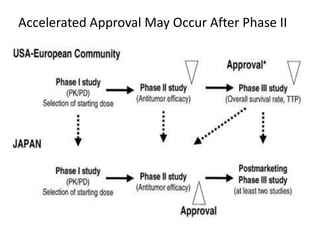
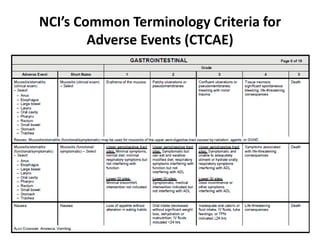
This document summarizes the key elements and stages of clinical drug trials. It discusses the rationale for conducting clinical trials to evaluate safety and efficacy of new drugs. Preclinical testing in cells and animal models is outlined. The main types of clinical trials - Phase I, II, and III - are described in terms of their objectives, patients, designs, and endpoints. Key components of a clinical trial protocol including background, objectives, eligibility criteria, treatment plan, response criteria, and statistical analysis are also summarized.