PHASE II CLINICALTRIALS
• Therapeutic exploratory trials
• Studied first time on patients with targeted disease
• Parameters
Therapeutic efficacy
Effective dose range
Safety revaluation and further pharmacological data
• Study population
100 to 300 patients
• Study design
Randomised controlled multicentric trial
Test drug is compared with standard or placebo
Single blinded - IIa
Double blinded - IIb( patient code with third party)
• Duration : Months to years.
• Dose - Usually, (but not always)less than the highest dose used in Phase I
• Study done by clinical investigator
• Usually drug development failure occur in this phase due to 1.lack of efficacy or 2.Toxic
effects/ADR.
2.
• II aPilot clinical trials to evaluate safety, efficacy and dose range in selected pts.
Population : 20 -200
Blinding : Single blinded study.
Parameters: Safety, efficacy
Dose range
Frequency of dosage
• II b Most vigorous demonstration of medical efficacy
Population :50 to 300
Blinding : Double blinded study
Study design used
1.Parallel group design
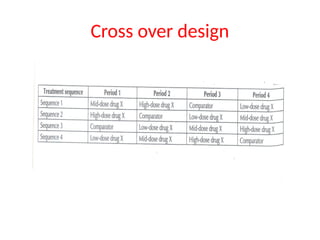
2.Cross over design
3.Factorial design
4.Group sequential design
3.
PARALLEL GROUP DESIGN
•Most commonly used one.
• Subjects allotted to 2 or more arms
• Each arm receive different drugs
(to be continued throughout the treatment)
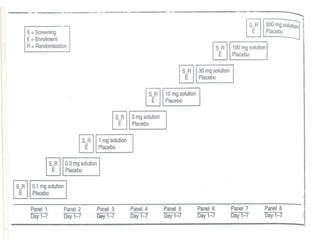
• One variation is ascending dose study
FACTORIAL DESIGN
• Simplestone is 2 x 2 factorial design
Group I – received drug A alone.
Group II – received drug B alone
Group III – received drug A and B
Group IV - received drug not A,not B.
• To study interaction between A and B.
7.
MODEL FORMAT PROTOCOLFOR PHASE II
• Title
• Objectives of study
• Research hypothesis
• Rationale of study
• Monitor/principal investigator/institutional review board
• Experimental design
• Patient population/inclusion,exclusion criteria
• Formation of standard and test dose
• Informed consent
• Pre study screening
• Study procedures
• Treatment period
• Follow up period
• Clinical lab investigations
• End of study visit
• End points
8.
TITLE
Randomised,multicentric,double blinded,active controlled,parallelarm
study of the oral factor Xa inhibitor in subjects undergoing elective total
knee replacement surgery.
OBJECTIVES OF STUDY
Primary objective is to determine the dose response and efficacy of oral
test drugs on the composite end point of adjudicated venous
thromboembolic events(VTE).(Asymptomatic and symptomatic DVT and
non fatal PE)and all cause death in subjects treated for 12 2 days
following elective unilateral total knee replacement surgery.
RESEARCH HYPOTHESIS
The research hypothesis is that test drug – direct and selective inhibition
of coagualtion factor Xa administrated orally once daily elicits dose
dependant reduction in VTE’s. Asymptomatic and symptomatic DVT and
non fatal PE and all cause death in subjects treated for 12 2 days
following elective unilateral total knee replacement surgery.
9.
RATIONALE FOR THESTUDY
Currently available anticoagulants are limited by a narrow therapeutic index or
multiple interactions with food and drugs or need frequent monitoring and dose
adjustment or need for parenteral administration.Test drug is an orally
active,selective,direct inhibitor of Xa.Safety and tolerability have been
demonstrated in Phase I healthy volunteer studies upto 7 days duration and these
studies have provided data to establish PK and PD profile of test drug.
The sponsor’s medical monitor is Name xxx. Address Madurai.
Principal investigator is Doctor yyy, M.D
Institutional review board is zzz, Madurai.
Experimental Design
Randomised,parallel arm , double blind ,active controlled multicentric study.
Patient population
Males and females 18 – 90 years scheduled to undergo elective unilateral total
knee replacement surgery.
10.
INCLUSION CRITERIA
Subjects of18 – 90 years of age of non child bearing potential who are undergoing
elective total knee replacement surgery.
EXCLUSION CRITERIA
• Child bearing age group women
• On Oral, implanted, Injectable contraception
• Pregnancy and lactation.
• Concurrent diseases
• Abnormal liver,renal,coagulation parameters
• Post operative use of epidural, intrathecal catheters
• Severe hypertension
• Active bleeding disorders
FORMULATION
• Test drug : 5 mg oral tablets, dose – 5mg O.D
• Control : Dabigatron oral tablets,50 mg B.D
INFORMED CONSENT
11.
PRE STUDY SCREENING
Upto30 days prior to study
Medical and social history
Physical examination and vital signs
Body weight,height,BMI
12 lead ECG
Liver and renal parameters
2 to 14 days before surgery
Hematology ,coagulation , INR and Aptt
Serum chemistry, urine analysis
STUDY PROCEDURE
Test drug 5 mg PO started on the evening of the day of surgery once a day every
evening 122 days.
Standard drug Dabigatron 50 mg on the evening of the day of surgery and 50 mg
B.D for 122 days.
(Parallel arm study)
12.
TREATMENT PERIOD –122 DAYS
Evaluate for symptomatic VTE’s (DVT and PE)
Bleeding events,AE’s,vital signs,INR and chemical parameters.
Bilateral ascending contrast venogram(primary end point)on day 122
Asymptomatic DVT should be treated as per protocol.
FOLLOW UP PERIOD
Day 122 to day 427
Evaluate for AE’s including symptoms suggestive of DVT and PE.
END OF STUDY VISIT
427 i.e, 307 from last day of treatment.
Do complete physical examination, clincal, lab parameters, coagulation profile,
urine analysis.
13.
END POINTS
PRIMARY EFFICACYEND POINT i.e, a composite of adjudicated VTE events
(asymptomatic and symptomatic DVT and non fatal PE)and all cause death in
subjects of treated for 122 days.
SECONDARY END POINTS
Non fatal or fatal MI
Coronary revascularisation procedures
Hospitalisation for heart failure
Non fatal or fatal stroke
All causes of mortality
THE PRIMARY SAFETY END POINT IS BLEEDING.














































































![CTEV [ clubfoot] DR ARUN LAL ,DR MOHAMED ASHRAF travancore medical college k...](https://cdn.slidesharecdn.com/ss_thumbnails/ctevclubfootdrarunlaldrmohamedashraftravancoremedicalcollegekollamkeralaindia-260208063247-18fc466c-thumbnail.jpg?width=640&height=640&fit=bounds)
