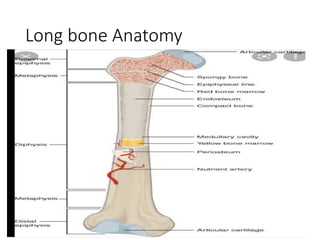
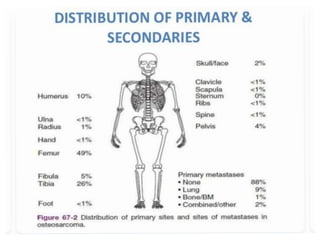
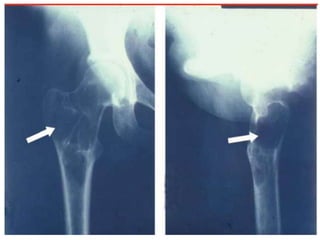
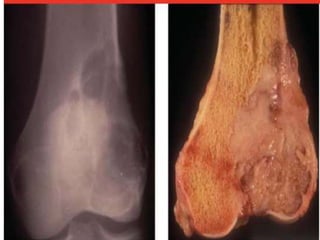
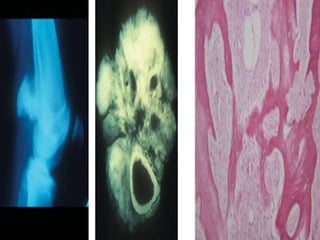
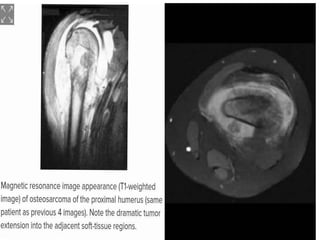
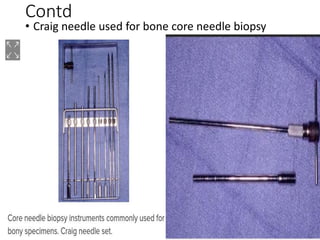
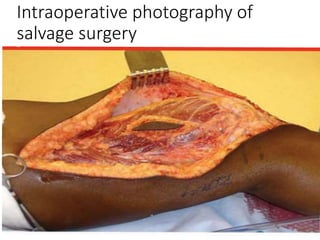
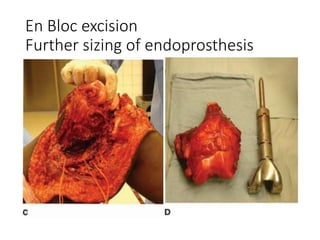
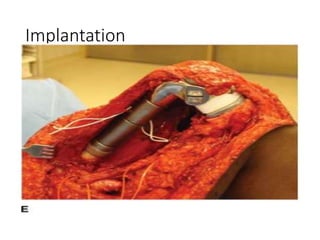
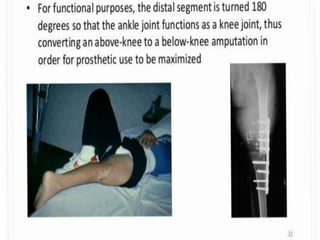
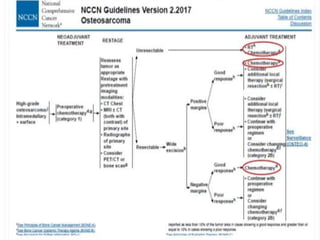
This document provides an outline and overview of osteosarcoma, including its epidemiology, pathology, clinical presentation, investigations, treatment, and prognosis. It begins with an introduction to osteosarcoma and outlines its surgical importance. Key points include that osteosarcoma most commonly affects the long bones of teenagers and young adults, neoadjuvant chemotherapy and limb salvage surgery have improved outcomes, and prognosis depends on several risk factors like tumor size and response to pre-op chemotherapy.







































































![References
• Meyers PA, Schwartz CL, Krailo M,et al: Osteosarcoma: The
addition ofmuramyl tripeptide to chemotherapy improves
overall survival: A report fromthe Children’s Oncology Group. J
ClinOncol 2008;26:633-638
• Gorlick R, Janeway K, Marina N. Osteosarcoma. Pizzo PA,
Poplack DG, eds. Principles and Practice of Pediatric Oncology.
7th ed. Philadelphia: Wolters Kluwer; 2016. 876-97
• Orthobullets
• Kim SY, Helman LJ. Strategies to Explore New Approaches in
the Investigation and Treatment of Osteosarcoma. Cancer
Treat Res. 2010. 152:517-528. [QxMD MEDLINE Link].](https://image.slidesharecdn.com/pathologyandosteosarcoma-230129161059-3669578a/85/Pathology-and-Osteosarcoma-pptx-88-320.jpg)















































![Osteosarcoma[2]](https://cdn.slidesharecdn.com/ss_thumbnails/osteosarcoma2-130423123803-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



















