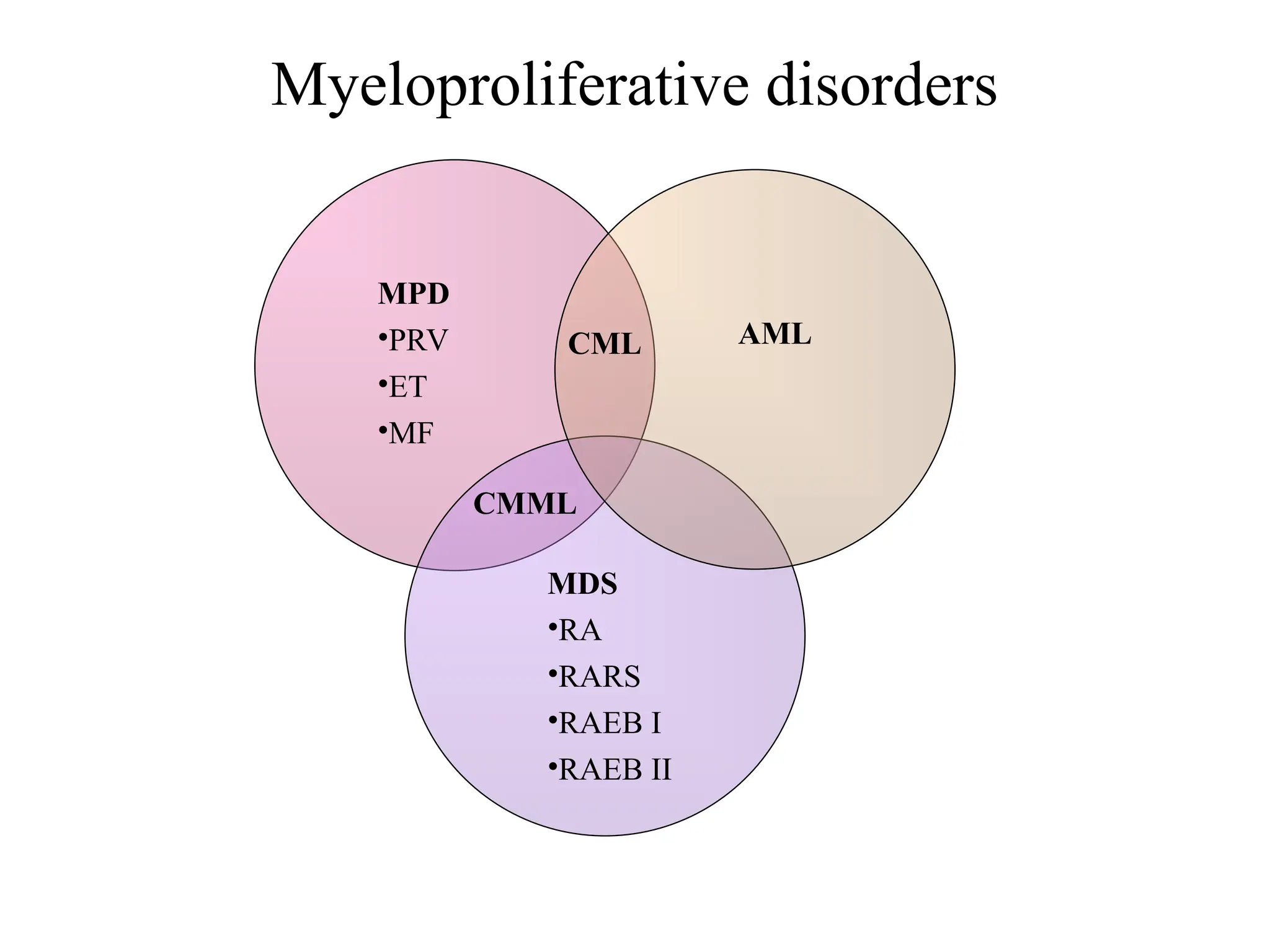
Myeloproliferative disorders
• ChMyeloid leukemia (BCR-ABL positive)
• Polycythemia Vera
• Essential Thrombocythemia
• Myelofibrosis
– Specific clincopathologic criteria for diagnosis and
distinct diseases, have common features
– Increased number of one or more myeloid cells
– Hepatosplenomegaly
– Hypercatabolism
– Clonal marrow hyperplasia without dysplasia
– Predisposition to evolve
5.
Bone marrow stemcell
Clonal
abnormality
Granulocyte
precursors
Red cell
precursors
Megakaryocytes Reactive
fibrosis
Essential
thrombocytosis
(ET)
Polycythaemia
rubra vera
(PRV)
Myelofibrosis
AML
Chronic myeloid
leukemia
70%
10% 10%
30%
6.
Epidemiology of CML
•Median age range at presentation: 45 to 55 years
• Incidence increases with age
– 12% - 30% of patients are >60 years old
• At presentation
– 50% diagnosed by routine laboratory tests
– 85% diagnosed during chronic phase
7.
Ionizing radiation LatentPeriod
Atomic bomb survivors 11 years ( 2-25)
Ankylosing spondylitis pts 3.6 years (1-6)
No evidence of other genetic factors
Chemical have not been associated with CML
Incidence 1-1.5/100,000 population
Male predominance
Epidemiology of CML
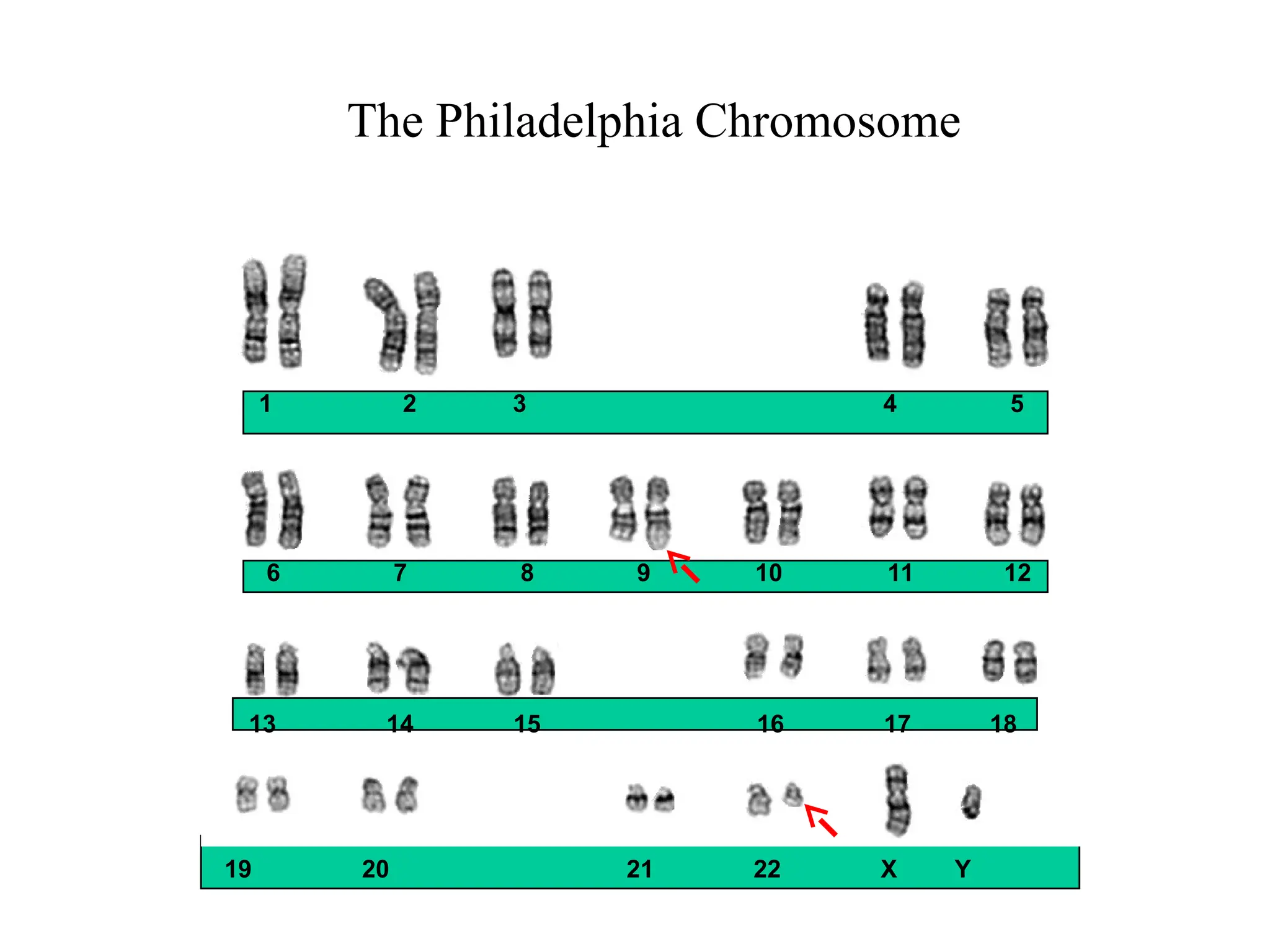
The Philadelphia Chromosome:t(9;22) Translocation
bcr-abl
Fusion protein
with tyrosine
kinase activity
22
bcr
abl
Ph
9 9+
Philadelphia
chromosome
12.
Clinical Course: Phasesof CML
Chronic phase
Median 4–6 years
stabilization
Accelerated phase
Median duration
up to 1 year
Blastic phase (blast crisis)
Median survival
3–6 months
Terminal phase
Advanced phases
13.
Treatment of ChronicMyeloid leukemia
Arsenic Lissauer, 1865
Radiotherapy Pusey, 1902
Busulfan Galton, 1953
Hydroxyurea Fishbein et al, 1964
Autografting Buckner et al, 1974
Allogeneic BMT (SD) Doney et al, 1978
Interferon Talpaz et al, 1983
Allogeneic BMT (UD) Beatty et al, 1989
Donor Leukocytes Kolb et al, 1990
Imatinib Druker et al, 1998
Imatinib/Combination therapy O’Brien et al, 200……
14.
CML Treatment
•Chemotherapy toreduce WCC - Hydroxyurea
•Interferon based treatment
•Allogeneic bone marrow transplant
•Molecular therapy - Imatinib
Issues related toBMT
• 70% long term cure rate
• Donor Availability
• Age of patient
• Length/stage of disease
• Treatment related mortality
• Long term sequalae – infertility, cGVHD
17.
The Ideal Targetfor Molecular Therapy
• Present in the majority of patients with a
specific disease
• Determined to be the causative abnormality
• Has unique activity that is
- Required for disease induction
- Dispensable for normal cellular function
18.
Mechanism of Actionof Imatinib
Goldman JM. Lancet. 2000;355:1031-1032.
Bcr-Abl
ATP
Substrate
Imatinib
Y = Tyrosine
P = Phosphate
Bcr-Abl
Substrate
P
P
P
P
19.
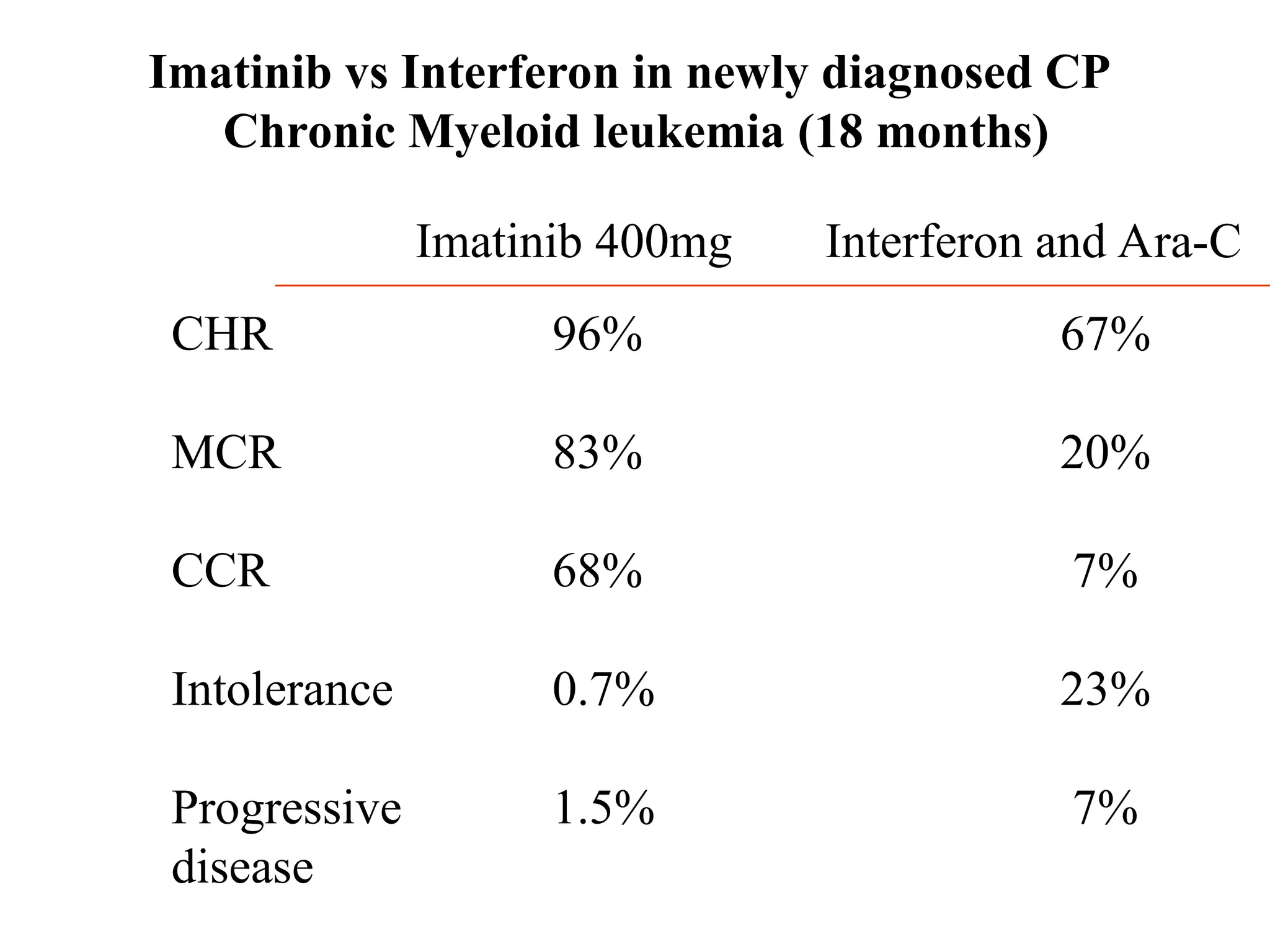
Imatinib compared withinterferon and low dose
Cytarabine for newly diagnosed chronic-phase
Chronic Myeloid leukemia
S.G. O’Brien et al
New England Journal of Medicine
Vol. 348 March 2003
Issues related toImatinib
• Very few molecular responses (5-10%)
• Resistance in some patients
• Lack of response in some patients
• Expensive
• Long term toxicity/side effects unknown
23.
CML
Diagnosis
Young with a
well-matcheddonor
Start Imatinib at
400mg/day
Cosider for Allograft
Allo SCT
Poor response or
Initial response
Followed by
Loss of response
Add or substitute
Other agents
Allo-SCT
Auto
Good response
maintained
Continue Imatinib
indefinitely
POLYCYTHEMIA VERA
• Chronic,clonal myeloproliferative disorder
characterized by an absolute increase in
number of RBCs
• 2-3 / 100000
• Median age at presentation: 55-60
• M/F: 0.8:1.2
27.
POLYCYTHEMIA VERA
JAK2 Mutation
•JAK/STAT: cellular proliferation and cell survival
• deficiency in mice at embryonic stage is lethal due
to the absence of definitive erythropoiesis
• Abnormal signaling in PV through JAK2 was first
proposed in 2004
• a single nucleotide JAK2 somatic mutation
(JAK2V617F mutation) in the majority of PV
patients
28.
Clinical features
• Plethora
•Persistent leukocytosis
• Persistent thrombocytosis
• Microcytosis secondary to iron deficiency
• Splenomegaly
• Generalized pruritus (after bathing)
• Unusual thrombosis (e.g., Budd-Chiari syndrome)
• Erythromelalgia (acral dysesthesia and erythema)
Diagnostic Criteria
A1 Raisedred cell mass
A2 Normal O2 sats and EPO
A3 Palpable spleen
A4 No BCR-ABL fusion
B1 Thrombocytosis >400 x 109/L
B2 Neutrophilia >10 x 109/L
B3 Radiological splenomegaly
B4 Endogenous erythroid colonies
A1+A2+either another A or two B establishes PV
32.
Treatment
• The mainstayof therapy in PV remains phlebotomy to keep the
hematocrit below 45 percent in men and 42 percent in women
• Additional hydroxyurea in high-risk pts for thrombosis (age over
70, prior thrombosis, platelet count >1,500,000/microL, presence
of cardiovascular risk factors)
• Aspirin (75-100 mg/d) if no CI
• IFNa (3mu three times per week) in patients with refractory
pruritus, pregnancy
• Anagrelide (0.5 mg qds/d) is used mainly to manage
thrombocytosis in patients refractory to other treatments.
• Allopurinol
33.
Essential Thrombocythaemia (ET)
•Clonal MPD
• Persistent elevation of Plt>600 x109/l
• Poorly understood
• Lack of positive diagnostic criteria
• 2.5 cases/100000
• M:F 2:1
• Median age at diagnosis: 60, however 20% cases <40yrs
34.
Clinical Features
• Vasomotor
–Headache
– Lightheadedness
– Syncope
– Erythromelalgia (burning pain of the hands or feet
associated with erythema and warmth)
– Transient visual disturbances (eg, amaurosis fujax,
scintillating scotomata, ocular migraine)
• Thrombosis and Haemorrhage
• Transformation
35.
Investigations
ET is adiagnosis of exclusion
• Rule out other causes of elevated platelet count
36.
Diagnostic criteria forET
• Platelet count >600 x 109/L for at least 2 months
• Megakaryocytic hyperplasia on bone marrow
aspiration and biopsy
• No cause for reactive thrombocytosis
• Absence of the Philadelphia chromosome
• Normal red blood cell (RBC) mass or a HCT <0.48
• Presence of stainable iron in a bone marrow aspiration
• No evidence of myelofibrosis
• No evidence of MDS
Activated protein Cresistance
• Activated protein C resistance
• Factor V leiden (R506Q) in 90% of cases
• Coagulation based assay (+/-FV def plasma)
• PCR based assay
• 2%-15%
• 2.0 –2.3% of Irish population are heterozygous FVL
Livingstone et al 2000
• 20% of unselected VTE
• Relative risk 3-8 fold for heterozygotes
Prothrombin G20210A
• Poort1996
• Mutation in 3’ UTR associated with
increased prothrombin levels
• 1.3% of Irish population heterozygous
(Keenan et al 2000)
• 6-8% of unselected VTE
• 16% of familial VTE
53.
Hyperhomocysteinemia
• Definite riskfactor for arterial vascular
disease
• >18.5 mol/l in 5% of normal population
• >18.5 mol/l in 10% of VTE
• Homozygous MTHFR (C677T) - 10% Irish
population
• Acquired B12, folate, B6 deficiency
54.
Antiphospholipid syndrome
• Venous,arterial or small vessel except
superficial venous thrombosis
• 3 consecutive unexplained fetal loss
• Severe pre-eclampsia or placental
insufficiency leading to prematurity (<34w)
• Unexplained single fetal loss >10 wks with
normal morphology
55.
APLS - laboratorydiagnosis
• ACL IgG or IgM (> 3SD above normal)
• Lupus anticoagulant
• Need 2 positive tests (either test will do) at
least 6 weeks apart
• Anti B2-Glycoprotein I
56.
Hormonal therapy
• OCPrisk of VTE increased x 2-3 fold (baseline
risk 1:10,000)
• FVL risk of VTE increased x 3-7 fold
• OCP + FVL risk of VTE increased x 33 fold
(30:10,000 = 0.3%)
• Need to screen 2 million to save one life
• Similar synergistic interaction with other
thrombophilic defects
• HRT likely to be similar
57.
Pregnancy and Virchow’striad
• Venous stasis - changes in tone and
obstruction
• Vascular damage at time of delivery
APTT, PS (free and total), APCr
FVIII:C, VWF, Fibrinogen
PAI-1 and PAI-2
58.
Pregnancy and venous
thromboembolicdisease
• Pregnancy increases risk x 5-10 fold
• 0.86/1000 deliveries
• 0.71/1000 (DVT) : 0.15/1000 (PE)
• Left leg >80%
• Ileofemoral more common than calf vein
(72% versus 9%)
• Increased with age, caesarian section, bed
rest and prior history of DVT/PE
59.
Clinical practice –DVT/PE
• Diagnosis
DVT – doppler ultrasound primarily (venogram gold
standard)
PE – ventilation perfusions scan primarily (pulmonary
angiogram is gold standard)
• Treatment
Heparin x 5-10 days until at least 5 days of warfarin
Warfarin x 6 months ( indefinite for second thrombosis)