X-linked adrenoleukodystrophy (X-ALD) is a genetic metabolic disorder that causes the breakdown of myelin in the brain. It is caused by mutations in the ABCD1 gene which prevents the breakdown of very long chain fatty acids. This leads to the accumulation of fatty acids in the brain and adrenal glands. X-ALD manifests in several forms from mild to severe. The severe childhood cerebral form can cause neurological deterioration leading to coma and death. There is no cure for X-ALD but treatments can help manage symptoms.
oALD is dueto the demyelination
of the white matter of the brain.
oIt is caused by a mutation in the
ABCD1 gene.
oALD takes several forms which
vary very widely in their severity
and progression.
5.
oIn patients withALD Brain
function declines.
oNeurons cannot conduct action
potentials.
oThey stop telling the muscles
what to do.
oAccumulation of very long chain
fatty acid (VLCFA).
oAbnormal immune response.
6.
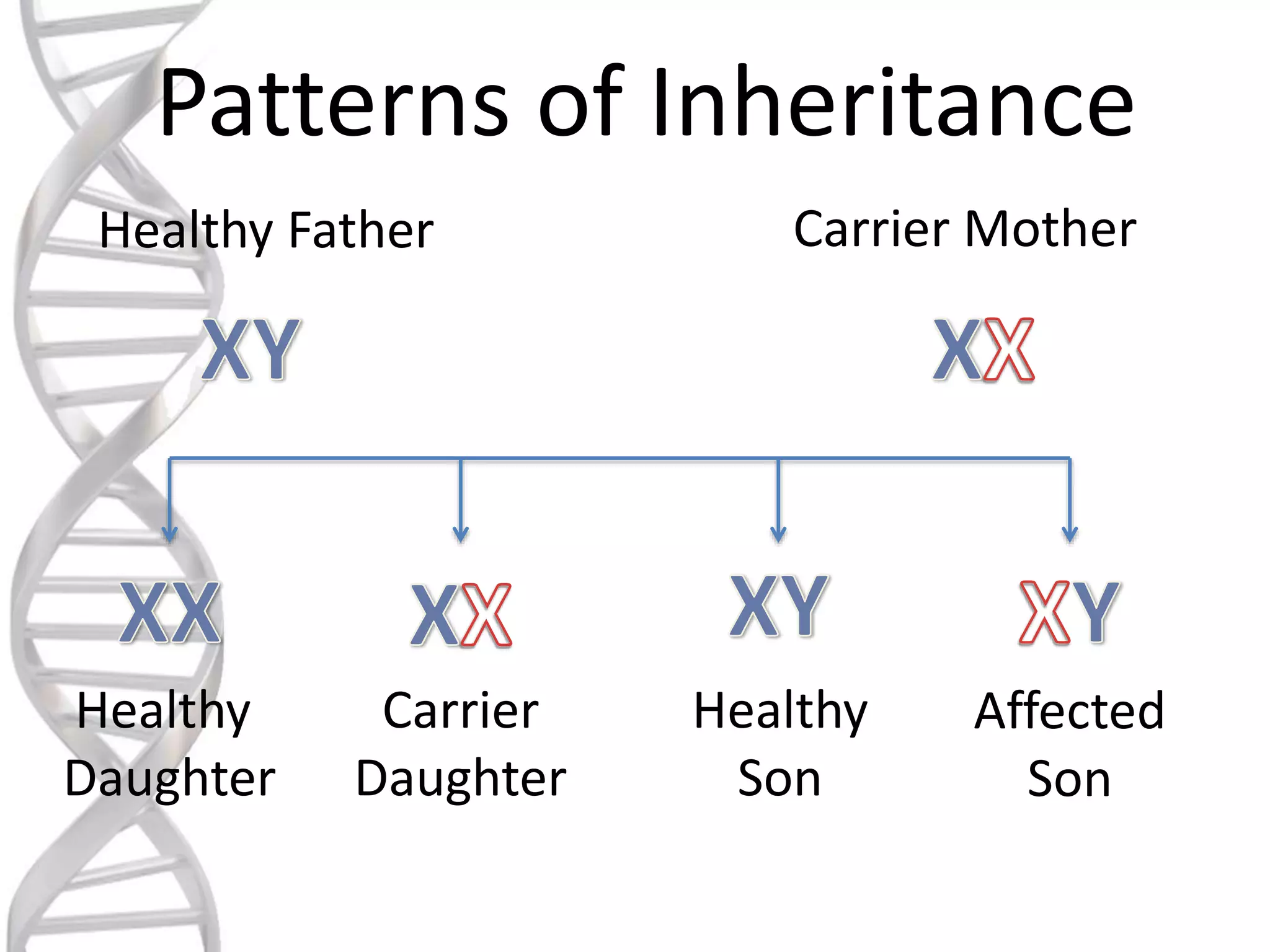
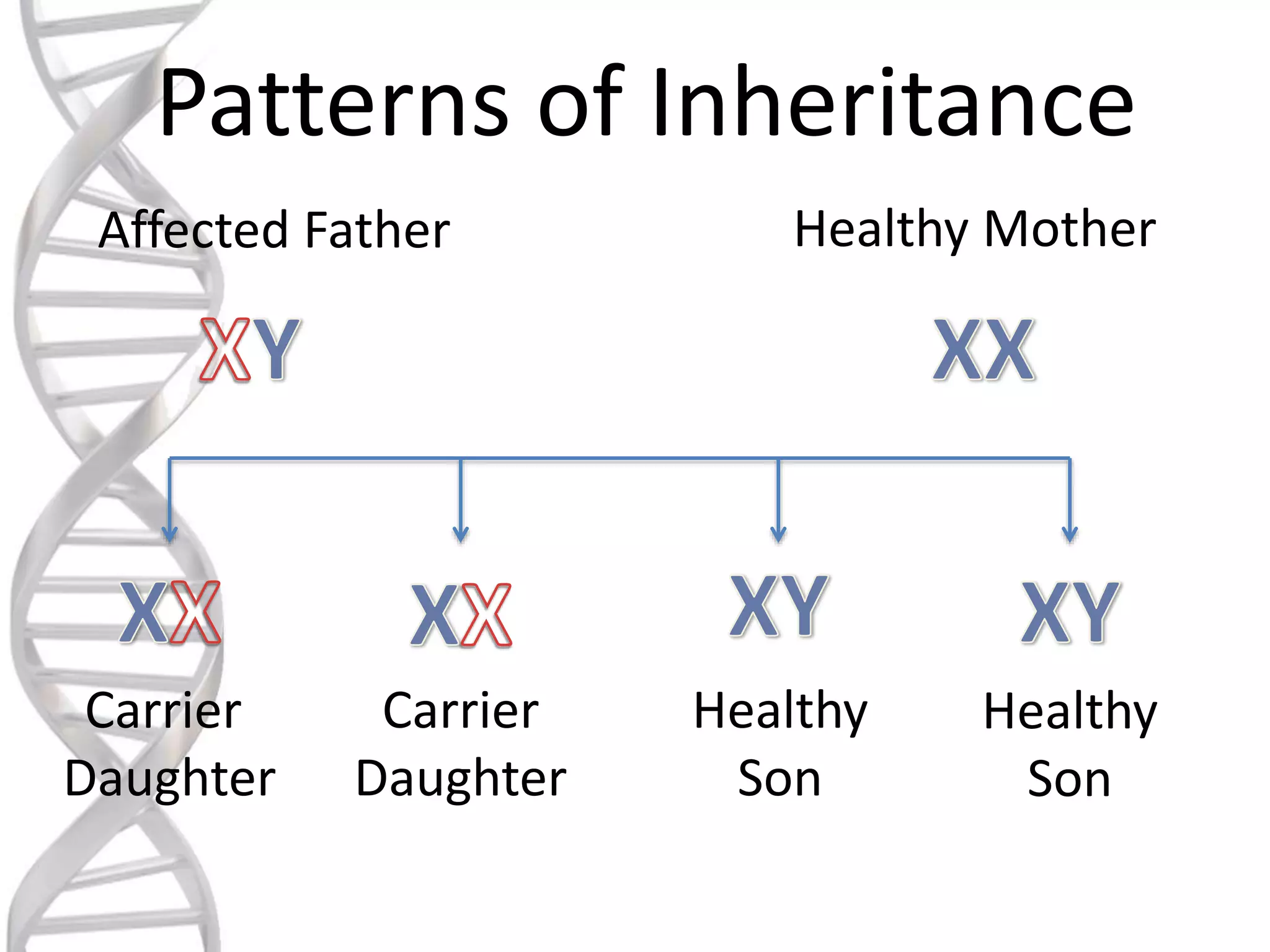
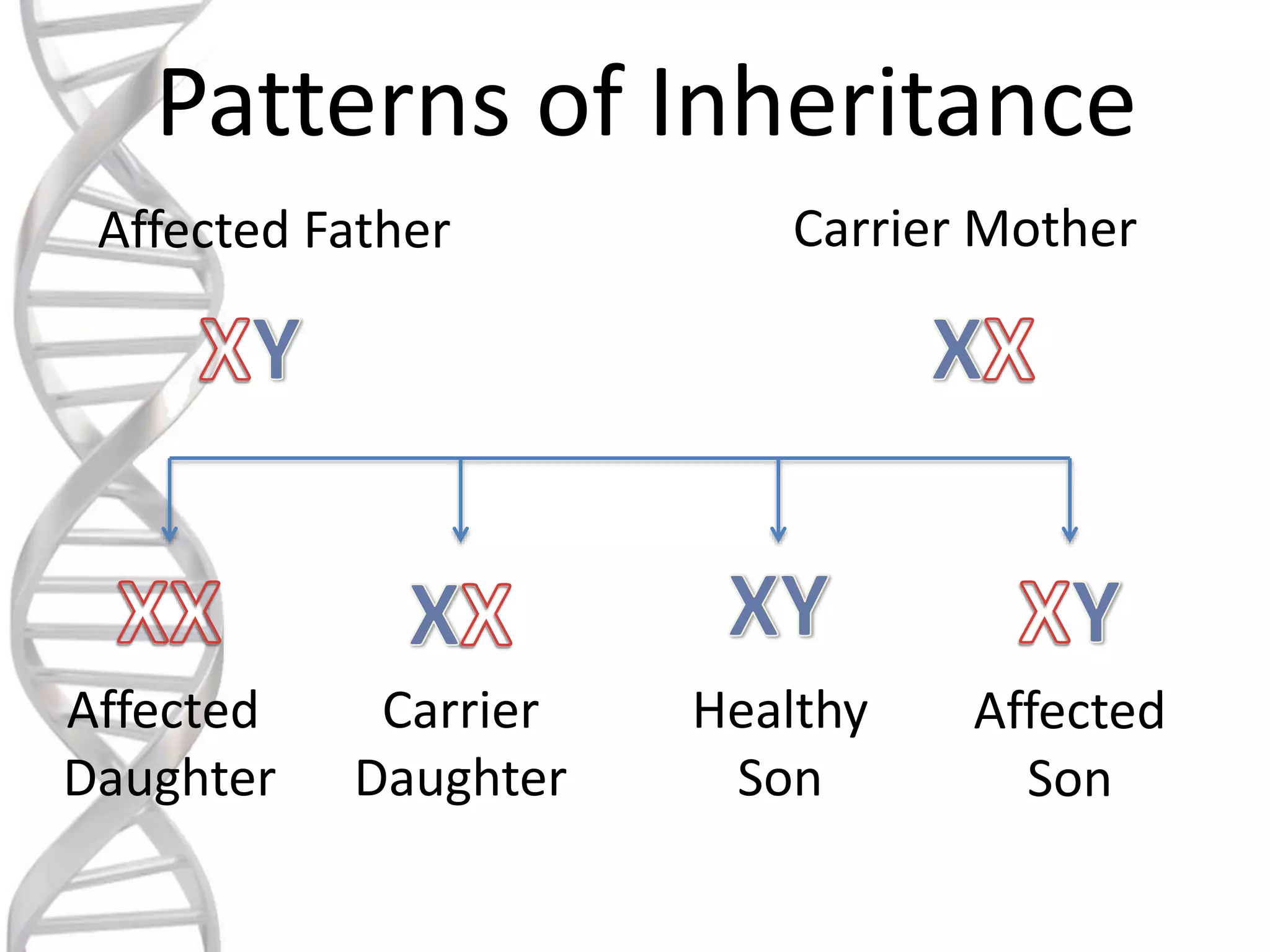
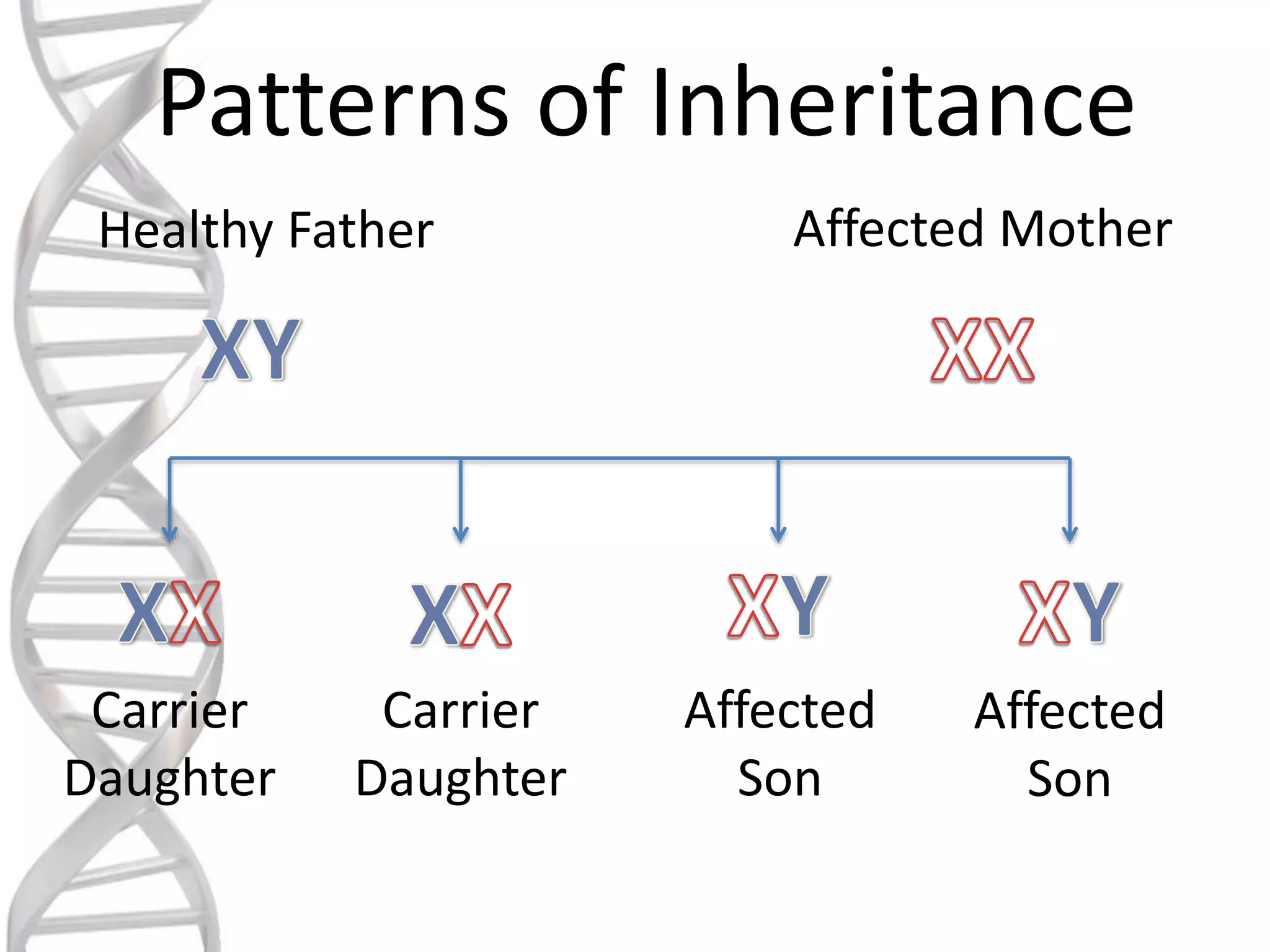
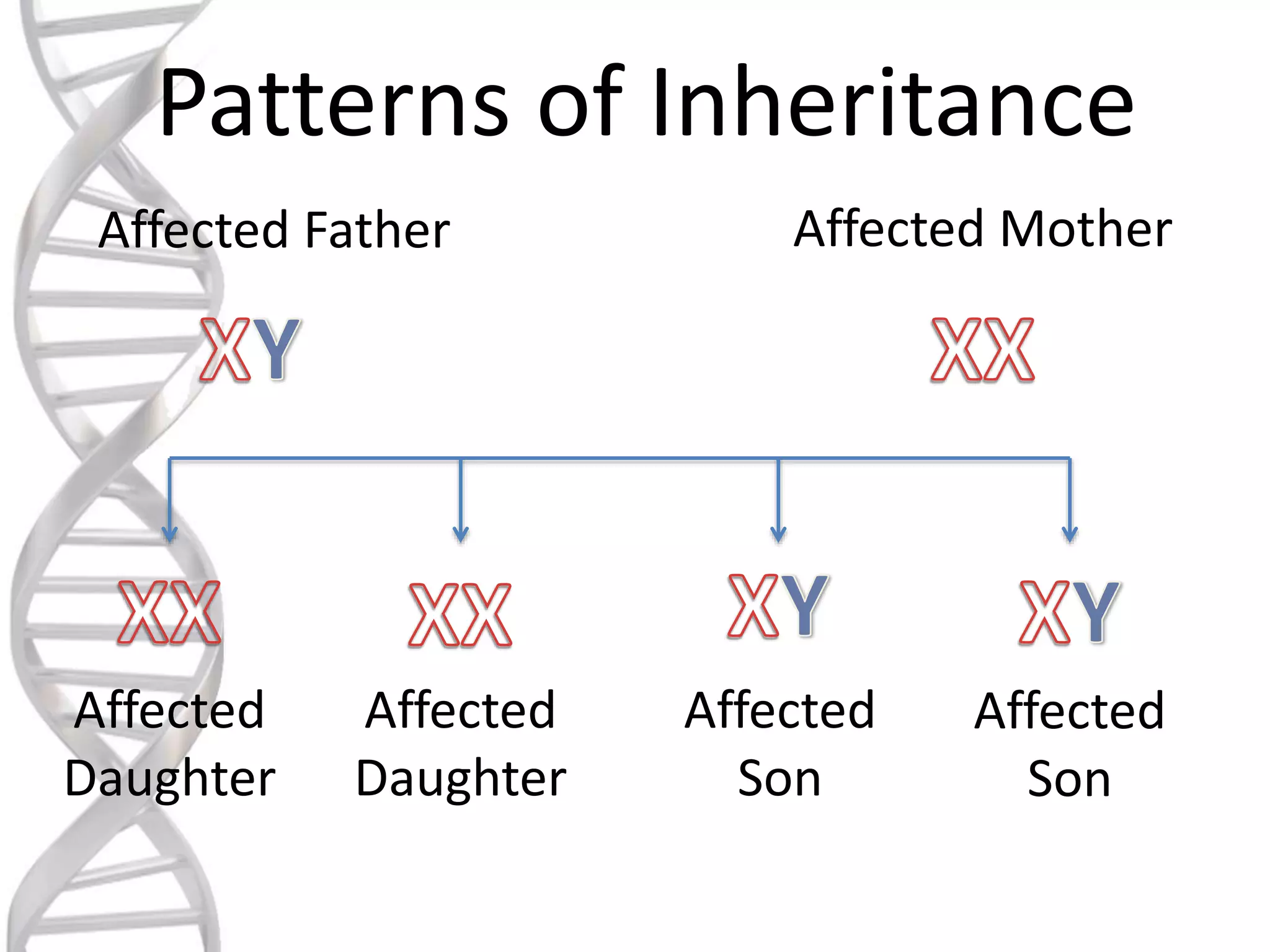
oALD is avery deadly genetic
disease that affects 1 in 18000
people.
oIt mostly affects boys and men.
oIt is a x-linked recessive
disorder.
oThis brain diseaseknows no
race or ethnics, it could affect
anyone.
oThe most devastating form of
ALD appears in childhood.
oWhere normal boys suddenly
show abnormal behavioral
problems.
13.
oThe symptoms growworse
(blindness, deafness, seizures,
loss of muscle control and
progressive dementia).
oLeads to permanent disability.
14.
o Symptoms ofALD vary from
person to person.
o With time patients are bedridden,
coma and eventually death
occurs.
15.
o If thepatient is treated and
taken care of well, the life span
can be extended for a few
years.
o But the patient might remain in
a devastating state, until death
arrives.
16.
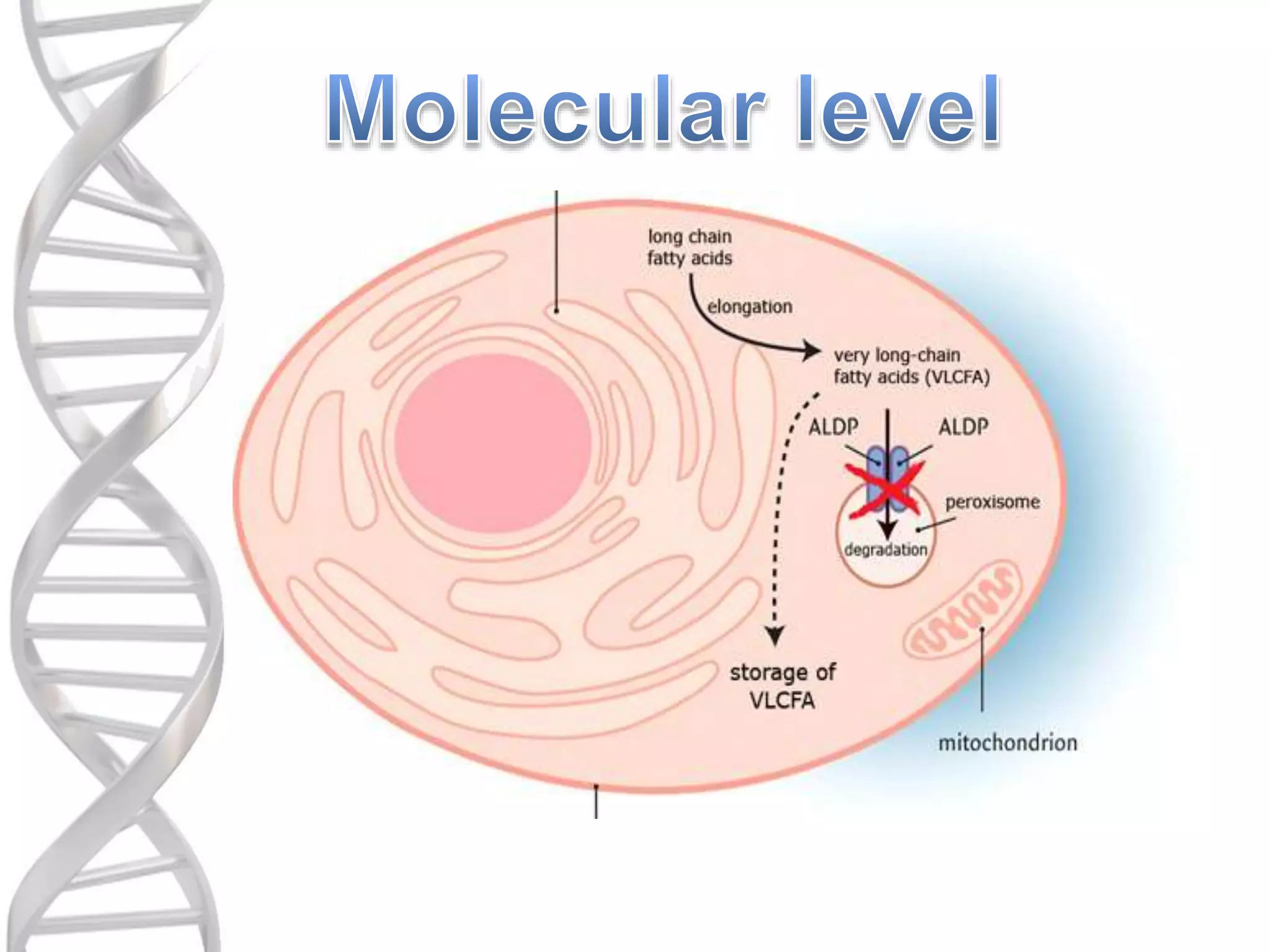
o ALD iscaused by a mutation in the
ABCD1 gene which is located on Xq28.
o ABCD1 (ATP binding cassette subfamily
D member 1) is the only gene that
encodes for ALD.
o It provides instructions for producing
Adrenoleukodystrophy protein (ALDP).
17.
oALDP is thoughtto be synthesized on
free polysomes, post-translationally
transported to peroxisomes, and inserted
into the membranes.
o It is involved in the transport of VLCFA
(Very long chain fatty acids) or VLCFA-
CoA (fatty acyl-CoAs) into the
peroxisomes.
18.
o Peroxisomes areorganelles that contain at
least 50 different enzymes.
19.
oA major functionof the peroxisome is the
breakdown of very long chain fatty
acids through beta-oxidation.
oThe long fatty acids are converted to
medium chain fatty acids, which are
subsequently shuttled to mitochondria
where they are eventually broken down
to carbon dioxide and water.
20.
oMutations in ABCD1interrupt the
production of ALDP.
oWithout ALDP, VLCFA are not
transported into and processed in
peroxisomes.
oTherefore they accumulate in glial
cells including oligodendrocytes.
21.
oUnlike most fattyacids, VLCFAs are
too long to be metabolized in
the mitochondria, and must be
metabolized in peroxisomes.
oIncorporation of VLCFA in myelin
destabilizes it causing it to break down
oVLCFA accumulation in the adrenal
cortex causes adrenal atrophy.
22.
oALDP deficiency andVLCFA
accumulation activate microglia
and initiate an inflammatory
reaction that further damages
myelin.
24.
o Although itall is due to the same
pathology ALD is expressed in different
forms.
o At first, this variation was thought to be
due to the nature of mutation.
o Recent researches suggest that a
modifier gene might account for some
of the phenotypic variability.
o Environmental factors may also play a
role.
25.
ALD Phenotypes
o ALDhas a variety of phenotypes.
o Most phenotypes appear in males.
o Childhood cerebral, Adolescent
cerebral, AMN, Adult cerebral,
Addison’s disease only, Asymptomatic.
o Some of the forms lead to the others.
o The various phenotypes co-occur
frequently in the same kindred or
nuclear family.
26.
o ALD geneabnormality without
neurological or endocrine
abnormalities.
o All individuals with the ALD gene are
free of clinical symptoms for at least the
first three years of life.
o Some may continue to have no
symptoms.
o The percentage of asymptomatic men
and women decreases with age.
27.
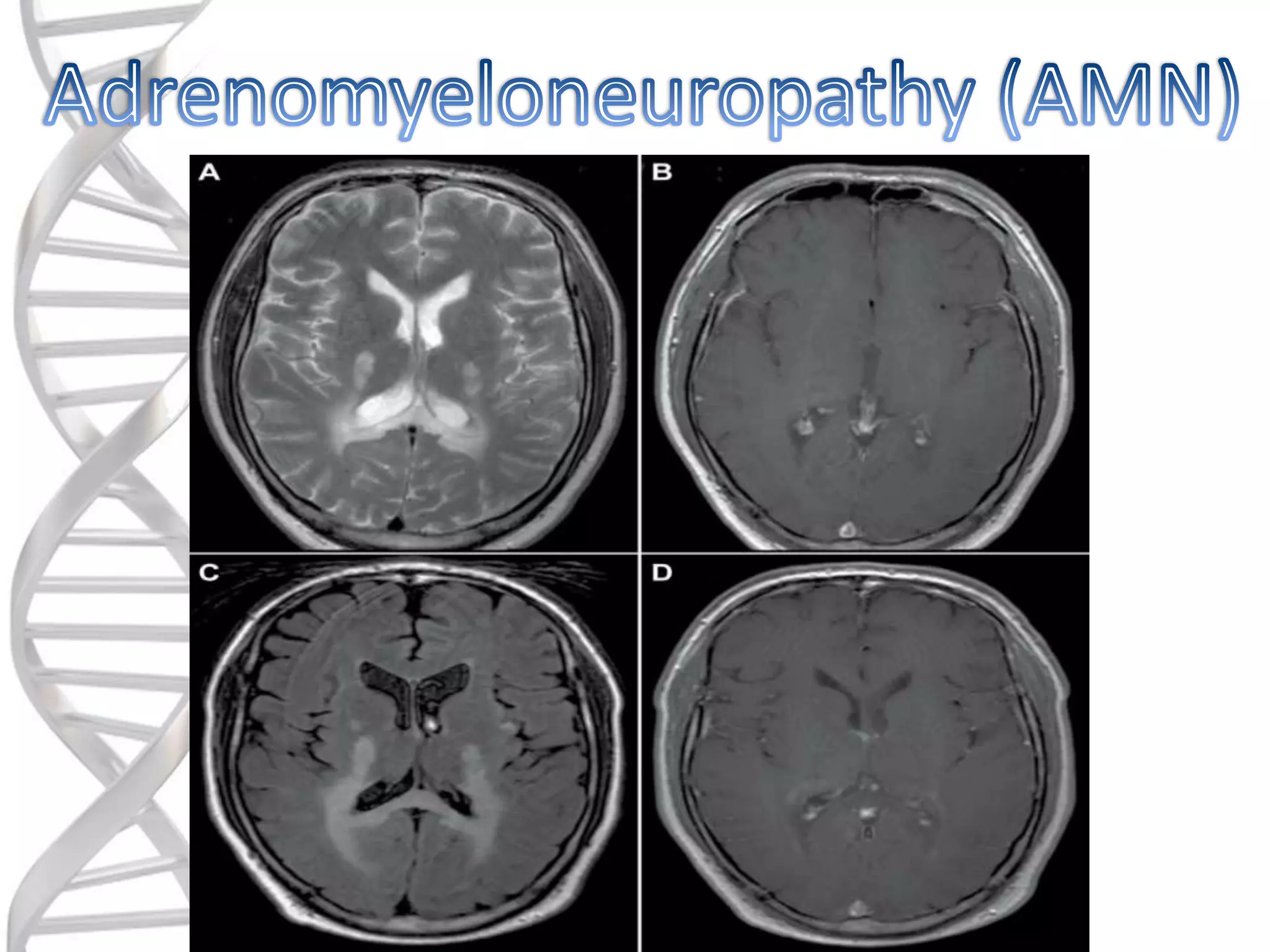
o is aninherited condition
that affects the spinal cord.
o which leads to the initial
symptoms that include
difficulties in walking or a
change in the walking
pattern.
28.
o The mostcommon form.
o Comprises about 40% of all patients.
o Virtually all patients who reach
adulthood develop AMN.
o Usually affects people at ages 20-30.
o Cerebral involvement later in 45% of
cases.
o Endocrinologic & Neurologic symptoms
29.
o General legweakness and stiffness
progresses into walking difficulty and
reduced balance. The use of mobility
devices may become necessary.
o Pain, numbness, or tingling in the legs.
o Weakness of the arms/hands.
o attacks of nausea, and generalized
weakness.
o Thin and scanty scalp hair, or balding.
30.
o Urinary problems,bowel urgency or
incontinence.
o Sexual dysfunction, or the inability to
obtain or maintain an erection.
o Weight loss.
o Cognitive defects, emotional
disturbances, and depression.
o Problems with thinking speed and
visual memory.
32.
o Is aninherited condition in which
the myelin sheath of the brain is
degenerated.
o This causes behavioral and
mental problems.
33.
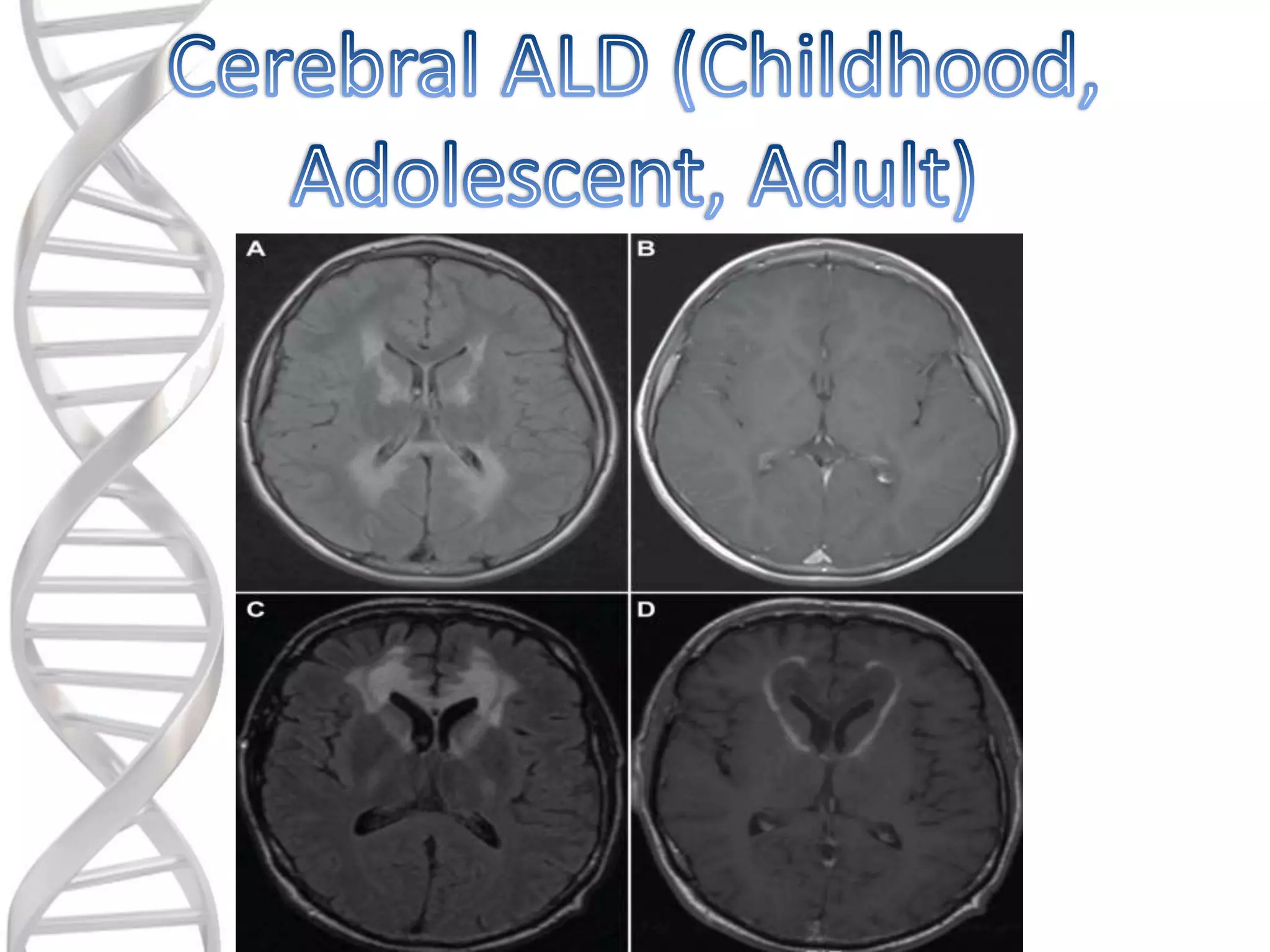
o These arethe most rapidly progressive
and devastating phenotypes of X-ALD.
o Childhood Cerebral makes up about
30% of all cases .
o Adolescent Cerebral makes up about
5% of all cases .
o Adult Cerebral makes up about 3% of
all cases.
34.
o Childhood Cerebralusually affects
boys between 4-10 years of age.
o Adolescent Cerebral usually affects
males between 11-21 years of age.
o Adult Cerebral usually affects
people from the age of 21 and older.
35.
o Behavioral problems& Hyperactivity.
o Lethargy, tires easily, clumsiness.
o Hypoglycemia
o Eye pain, double vision, visual
problems, blindness.
o Hearing loss.
o Tanning or bronzing of the skin
o Crossed eyes.
36.
o Attention deficitdisorder (ADD)
o Difficulty reading & understanding
written material.
o Difficulty understanding speech.
o Swallowing difficulties.
o Adrenal insufficiency.
o Recurring viral infections.
o Seizures.
37.
o Changes inmuscle tone, especially
muscle spasms and uncontrolled
movements.
o Worsening nervous system damage,
including coma, decreased fine
motor control, and paralysis.
39.
Is a conditionin which the adrenal
glands do not produce adequate
amounts of steroid hormones, primarily
cortisol; but may also include impaired
production of aldosterone (a
mineralocorticoid), which regulates
sodium conservation, potassium
secretion, and water retention.
40.
o adrenocortical insufficiencywithout
neurological abnormalities.
o About 10% of ALD are of this form.
o Both young boys and adult males
can be affected.
41.
o Coma
o Darkeningof the skin.
o Loss of weight and muscle mass
(wasting)
o Muscle weakness and fatigue
o Decreased appetite
o Nausea
o Abdominal pain
o Low blood pressure.
42.
o As inmany X-linked diseases, it was
assumed that female carriers remain
asymptomatic.
o However, many women develop AMN-
like symptoms.
o Symptoms appear later in life.
Diagnosing
Adrenoleukodystrophy
oYour doctor mayalso look for damage
to your brain using an MRI scan.
oSkin samples or a biopsy and
fibroblast cell culture can also be
used to test for VLCFAs.
45.
Treatment of
Adrenoleukodystrophy
o Thereis no cure for ALD but the severity
of the symptoms can be decreased.
o Treatment methods depend on the type
of ALD you have
o Steroids can be used to treat Addison’s
disease. There are no specific methods
for treating the other types of ALD.
46.
Treatment of
adrenoleukodystrophy
Some peoplehave been helped by:
o switching to a diet that contains low levels
of VLCFAs
o taking Lorenzo’s oil to help lower elevated
VLCFA levels
o taking medications to relieve symptoms
such as seizures
o doing physical therapy to loosen muscles
and reduce spasms
47.
Treatment of
Adrenoleukodystrophy
oDoctors continueto look for new ALD
treatments. Some doctors are
experimenting with bone marrow
transplants. If children with childhood
cerebral ALD are diagnosed early,
these experimental procedures may
be able to help.
48.
Preventing
Adrenoleukodystrophy
o Because ALDis an inherited condition,
there’s no way to prevent it. If you’re a
woman with a family history of ALD, your
doctor will recommend genetic
counseling before you have children.
An amniocentesis or chorionic villus
sampling can be done during pregnancy
to determine if your unborn child is
affected.
49.
Outlook of
Adrenoleukodystrophy
o Childhoodcerebral ALD can lead to
severe disability, coma, and death. Coma
typically occurs around two years after
symptoms begin appearing and can last
for up to 10 years, until death.
o Adrenomyelopathy and Addison’s disease
are not as serious as childhood cerebral
ALD. They progress at a slower rate. The
symptoms can be treated, but there is no
cure for ALD.
50.
Resources
o Wikipedia.org
o Semanticscholar.org
oStopald.org
o Medlineplus.gov
o ghr.nlm.nih.gov
o ncbi.nlm.nih.gov
o genomicseducation.hee.nhs.uk
o x-ald.nl
o myelin.org
o Ulf.org
o ojrd.biomedcentral.com
o pathlabs.rlbuht.nhs.uk
o neuropathology-web.org
o bscb.org
o rarediseases.info.nih.gov
o Healthcare.com