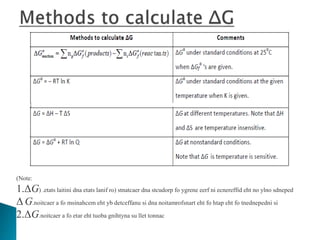
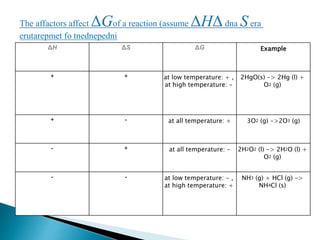
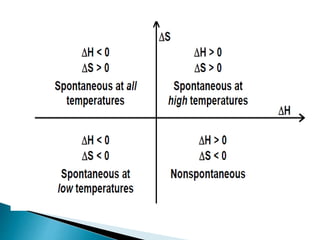
This document discusses Gibbs free energy (G) and how it relates to chemical reactions and equilibrium. It defines G and explains how it depends on enthalpy (H), entropy (S), pressure (P), and temperature (T). It describes how the change in G for a reaction (DG) determines whether the reaction is spontaneous. It shows how DG can be used to calculate equilibrium constants (K) at different temperatures and how K values change with temperature.

















![R is the gas constant (8.314 J/K•mol)
T is the absolute temperature (K)
Q is the reaction quotient = [Products]0 or P°Products
18.4
So we Can correct Gibbs Free Energy change for non-
standard state
[Reactants]0 P°Reactants
Q=reaction quotient indicating the state of a system at a given instant.
Conc.
Pressure](https://image.slidesharecdn.com/lec2dr-231104143341-b1e66c45/85/lec-2-dr-marwa-ppsx-19-320.jpg)




![2 NO (g) + O2 (g) 2 NO2 (g)
Calculate DG0 for the following reaction at 25 0C?
Given:
DH° S°
NO 90.37 kJ/mole 210.62 j/molK
O2 0 205.0
NO2 33.84 240.45
DG0 = DH0 – TDS0
DS0
rxn
= 2 x S0(NO2) – [2 x S0(NO) – S0(O2)]
DH0
rxn
= 2 x DH0(NO2) – [2 x DH0(NO) + DH0(O2)]
f
f f f
= 2(33.84 kJ) - 2(90.37 kJ) - 0 = -113.06 kJ
= 2(240.45J/molK) - 2(210.62 J/molK) - 205.0 j/molK
= -145.34 J/K](https://image.slidesharecdn.com/lec2dr-231104143341-b1e66c45/85/lec-2-dr-marwa-ppsx-24-320.jpg)
![DG0 = DH0 – TDS0
DG0
rxn
= 2 x DG0(NO2) – [2 x DG0(NO) + DG0(O2)]
f f
f
= 2(51.84 kJ/mol) - 2(86.71 kJ/mol) - 0
= 103.68 kJ - 173.42 kJ
= -69.74 kJ
= -113.06 kJ - (298 K)(- 0.14534 kJ/K
= -113.06 kJ + 43.31 kJ = -69.75 kJ
Can also calculate with DG0 ’s from tabulated data
f](https://image.slidesharecdn.com/lec2dr-231104143341-b1e66c45/85/lec-2-dr-marwa-ppsx-25-320.jpg)