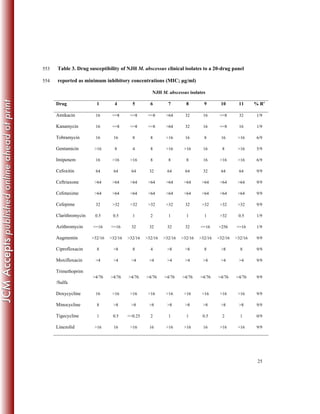
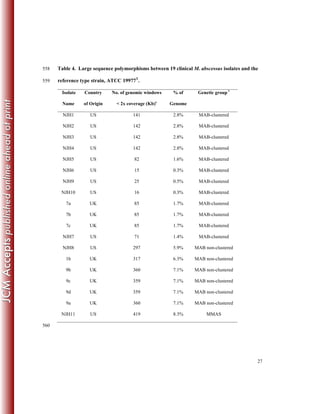
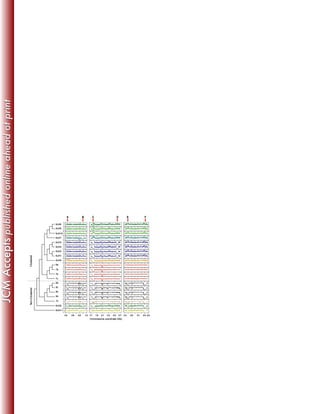
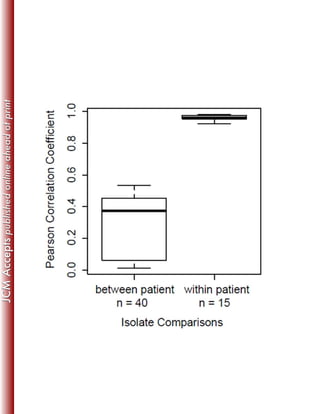
This study sequenced the genomes of 11 clinical Mycobacterium abscessus isolates from 8 US patients with pulmonary infections. Core genome analysis compared these isolates to 30 globally diverse strains to investigate population structure. Longitudinally sampled isolates showed very few genetic differences, suggesting homogenous infection populations. Genome content variation between isolates was 0.3-8.3% compared to the reference strain, indicating plasticity.









![9
Nucleotide Sequence Accession Numbers.180
SRX641283, SRX641284, SRX641291, SRX641292, SRX641293, SRX641294, SRX641295,181
SRX339602, SRX339603182
183
Results and Discussion184
Clinical and Microbiological Attributes of Patients and Isolates185
M. abscessus isolates examined in this study were obtained from sputum or bronchoalveolar186
lavage (BAL) samples from eight patients referred to National Jewish Health (NJH), in Denver,187
Colorado for management of chronic M. abscessus-related pulmonary infections between 2009188
and 2011 (Table 1). All but one patient had a ≥ 2-year history of M. abscessus pulmonary189
infection and all had received prolonged treatment with multiple anti-mycobacterial drugs190
including macrolides and aminoglycosides. Isolates NJH1 to NJH4 were sampled longitudinally191
from the same patient at three time points during a 6-month period; isolates NJH2 and NJH3192
were individual colonies collected at the second time point. All patients’ primary residences were193
in the northern or eastern United States, with the exception of one individual from Puerto Rico194
(isolate NJH7). Patients were primarily female (6/8), greater than 60 years old (7/8), and the195
majority of patients were smear negative (6/7) at the time of sputum collection. Four of the196
patients had adult cystic fibrosis (NJH1, 5, 8, 9) and two were carriers of a CFTR polymorphic197
allele (NJH7, 11).198
All isolates were initially identified to the sub-species level by Sanger sequencing of the199
rpoB and hsp65 genes. Based on sequence homology to type strains, isolates NJH1 to NJH10200
were identified as M. abscessus subsp. abscessus, and isolate NJH11 was identified as M.201
abscessus subsp. massiliense. All isolates were also evaluated for the erm(41) deletion [16]. In202](https://image.slidesharecdn.com/f34ee745-831d-455e-bb91-c7352390f46c-160414225358/85/J-Clin-Microbiol-2014-Davidson-JCM-01144-14-9-320.jpg)