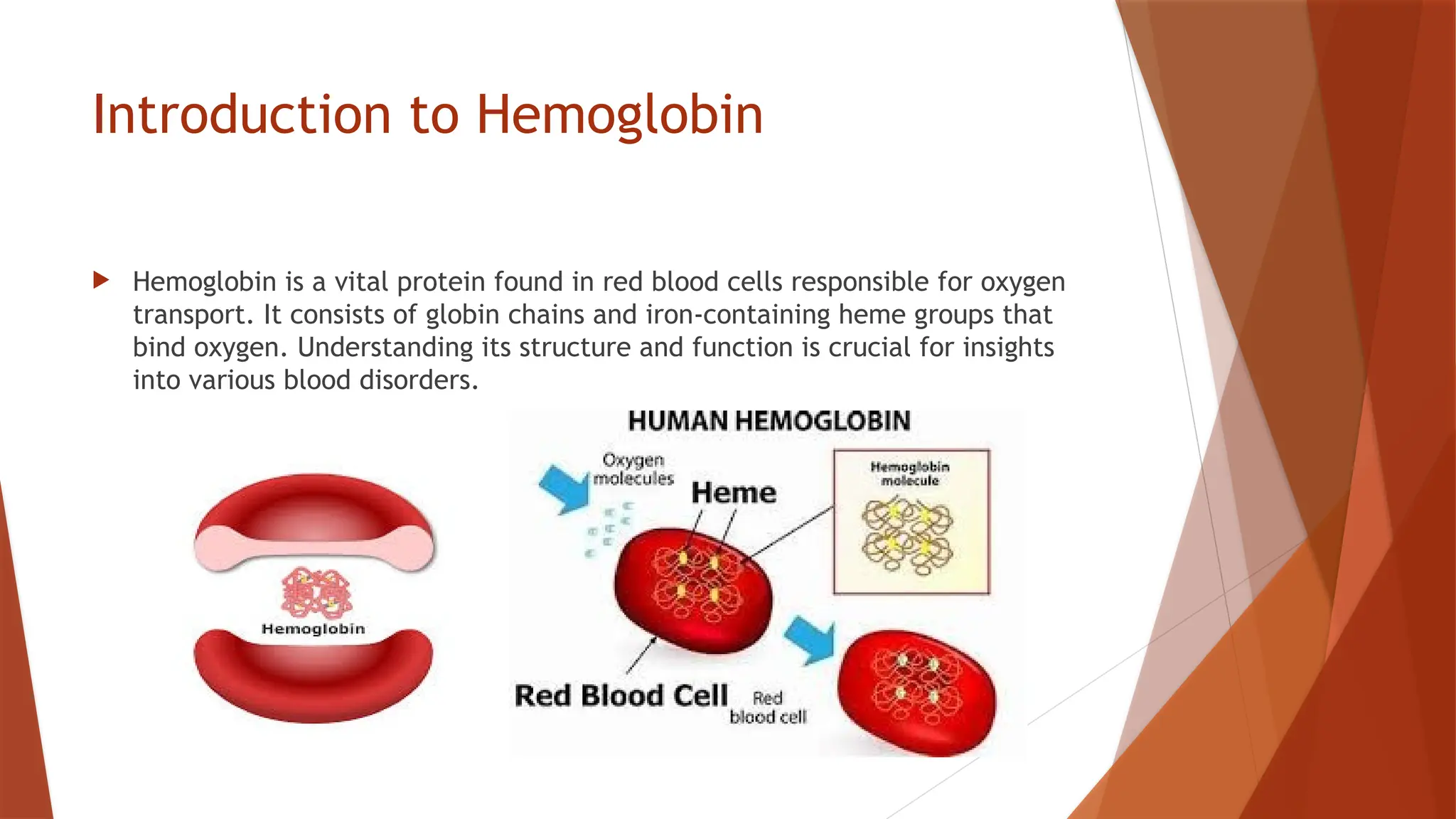
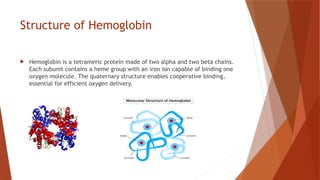
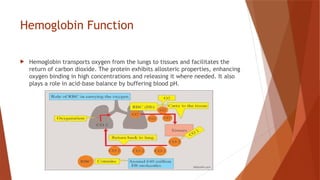
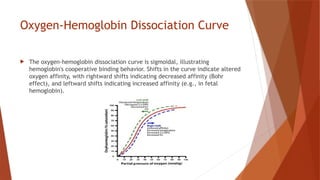
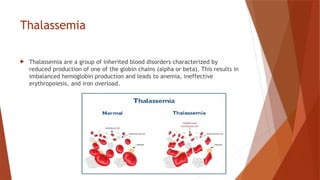
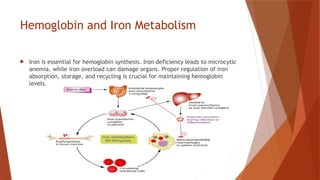
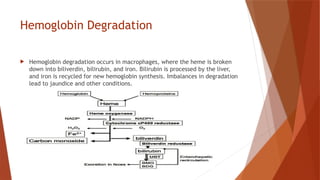
Hemoglobin is a critical protein in red blood cells that transports oxygen and consists of globin chains and heme groups. Its tetrameric structure allows for cooperative binding and plays a crucial role in various conditions, including sickle cell disease and thalassemia. Understanding hemoglobin's functionality and related disorders is essential for diagnosing and managing blood-related health issues.