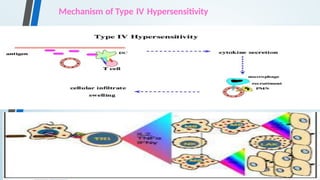
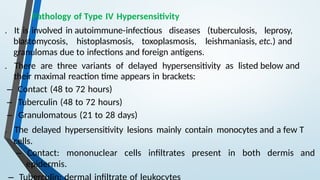
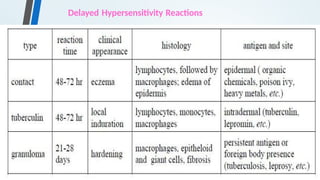
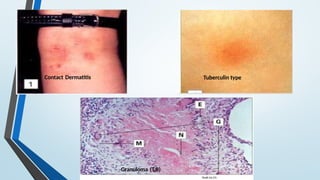
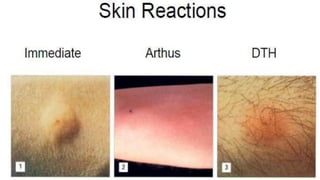
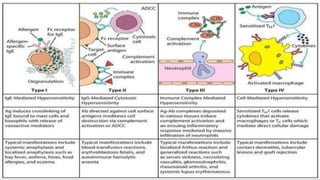
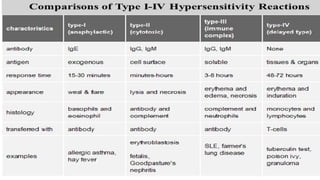
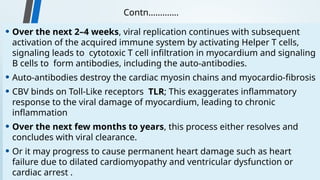
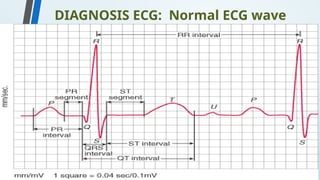
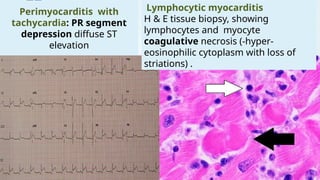
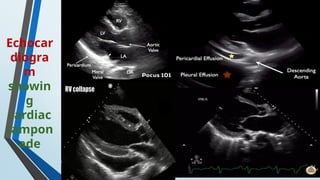
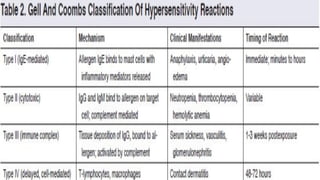
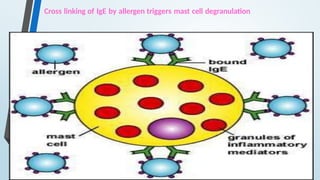
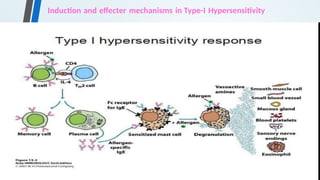
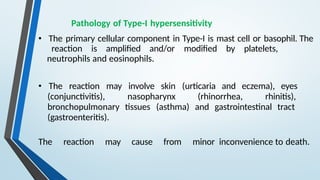
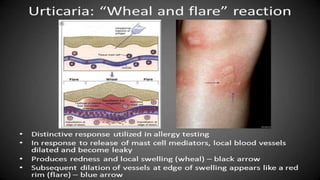
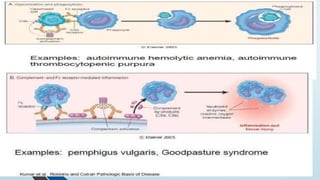
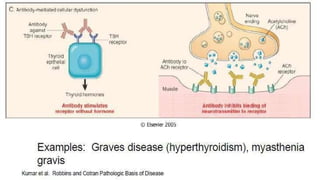
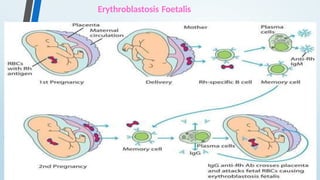
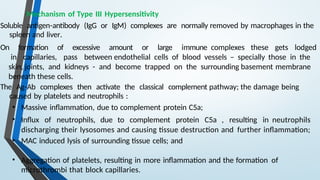
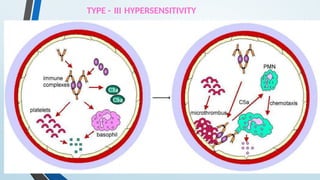
The document discusses hypersensitivity reactions categorized into four types (I-IV), with a focus on the mechanisms, characteristics, and pathology of each type. It details the immunological responses involved, such as immediate (type I) allergic reactions and delayed (type IV) cell-mediated responses, emphasizing their effects on the body and examples of related diseases. Furthermore, it explores specific responses and conditions like anaphylaxis, autoimmune diseases, and immune complex-mediated disorders.
























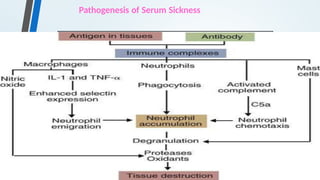
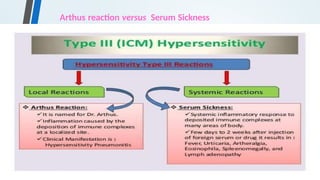
![Serum Sickness
• Serum sickness is a systemic type III hypersensitivity reaction that
results from the injection of heterologous or foreign protein or
serum.
• Certain medications (eg, penicillin, nonsteroidal anti-
inflammatory drugs [NSAIDs]) have also been associated with
serum sickness– like reactions.
• These reactions typically occur 1 to 3 weeks after exposure to the drug,
but may occur as early as 1 to 24 hours afterward.
• Clinical manifestations include general malaise, fever, urticaria,
artheralgia, eosinophilia, splenomegaly and lymphadenopathy.](https://image.slidesharecdn.com/hypersensitivityreactionsautoimmunediseases-241014054033-3f01705f/85/HYPERSENSITIVITY-REACTIONS-AUTOIMMUNE-DISEASES-pptx-36-320.jpg)