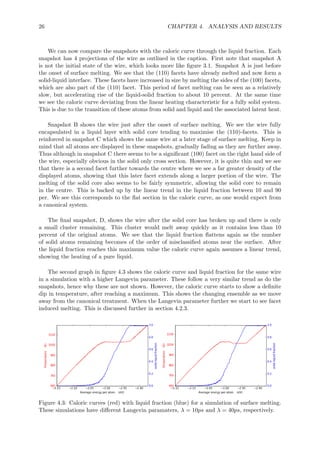
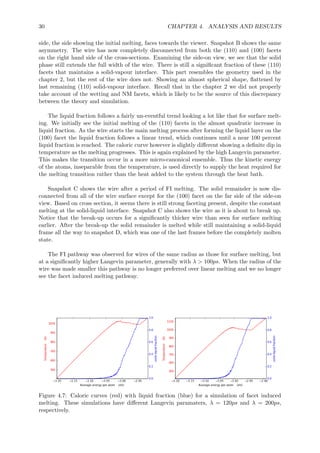
This document summarizes the different pathways a cylindrical metal nanowire can melt through: surface melting, linear melting, and facet induced melting. A simple model is developed based on free energy considerations to predict the preferred melting pathway based on the wire radius and interfacial energy differences. Molecular dynamics simulations of aluminum nanowires demonstrate the different pathways and confirm radius is an important parameter. The simulations also show the interface energies and shrinkage of non-melting facets influence the melting pathway.











































![[헬로! 언플러그드 1주차] 아바타 놀이](https://cdn.slidesharecdn.com/ss_thumbnails/1-161224100914-thumbnail.jpg?width=640&height=640&fit=bounds)

![[헬로! 언플러그드 3주차] 유한 오토마타 & 마무리](https://cdn.slidesharecdn.com/ss_thumbnails/3-161224100915-thumbnail.jpg?width=640&height=640&fit=bounds)




![[알고리즘 스터디 3주차]기수정렬/계수정렬/버킷정렬](https://cdn.slidesharecdn.com/ss_thumbnails/3radixcountingbucket-161127154723-thumbnail.jpg?width=640&height=640&fit=bounds)

![[알고리즘 스터디 2주차]병합정렬/퀵정렬/힙정렬](https://cdn.slidesharecdn.com/ss_thumbnails/2mergequickheap-161127154726-thumbnail.jpg?width=640&height=640&fit=bounds)

![[알고리즘 스터디 1주차]삽입정렬/버블정렬/선택정렬](https://cdn.slidesharecdn.com/ss_thumbnails/1inserttionbubbleselection-161127154727-thumbnail.jpg?width=640&height=640&fit=bounds)
![[알고리즘 스터디 4주차]LCS 알고리즘](https://cdn.slidesharecdn.com/ss_thumbnails/4lcs-161224045826-thumbnail.jpg?width=640&height=640&fit=bounds)

![[아두이노 워크샵 1차] 아두이노 소개 / LED / 피에조 부저 / 버튼](https://cdn.slidesharecdn.com/ss_thumbnails/1pdf-161224052608-thumbnail.jpg?width=640&height=640&fit=bounds)



















