This dissertation studies synthetic molecular motors and self-assembled monolayers through computational modeling and simulation. Chapter 1 provides background on molecular motors and the theoretical methods used, including density functional theory and classical force fields. Chapter 2 models a caltrop-based rotary motor driven by rotating electric fields, finding resonance effects that enhance friction. Chapter 3 presents a novel power law mechanism for dissipation in motors indirectly coupled to a thermal bath. Chapter 4 models the kinetics of monolayer displacement and finds it follows a universal perimeter-dependent growth model. Chapter 5 provides preliminary results on barriers to rotation in lanthanide double-decker complexes as potential rotary motors.
















![Chapter 1
Introduction
1.1 Molecular motors background
Molecular motors are molecular-scale machines that can convert different types of
input energy into mechanical energy, which is further used to perform useful work.
The fundamental difference between the macroscopic motors and the molecular-
scale ones is that the latter operate in an environment that is governed by thermal
fluctuations and they cannot move deterministically.
Nature first constructed tiny and elegant mechanical-like devices that use chem-
ical energy in order to perform numerous functions within cells, such as cell division
or intracellular transport. Although they operate in a world where Brownian mo-
tion and viscous forces dominate, biological motors are capable of achieving highly
controlled rotary and translational motion. For example, ATP-synthase is a rotary
motor that synthesizes the molecule adenosine triphosphate (ATP), which is the
energy currency within cells [1]. The motor rotates with discrete steps of 120◦
and generates a constant force of 40 pN, the highest value among reported mo-
tor proteins. The work done in one-third of a revolution is about 80 pN · nm or
just 20 times larger than thermal energy [2]. Cells also employ a variety of linear
motors that move along and exert forces on filamentous structures. For example,
kinesins and dyneins transport cargo along microtubules and myosins slide on actin
filaments generating forces up to 5-10 pN [3].
Inspired by biological motors, people started designing artificial molecular mo-
tors in attempts to understand and to augment basic motor functions.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-16-320.jpg)
![2
Artificial molecular motors can be classified according to the input energy they
use, the environment where they operate, or the type of mechanical motion they
produce. They use sources of energy such as: chemical, thermal, gas or liquid
flow, light, electric fields, etc. in order to produce linear or rotary mechanical-like
motion. The motors can operate immersed in liquids or gases, buried in solids, or
chemically attached to surfaces. The disadvantage of liquid and gas environments
is that the gas or liquid non-motor molecules apply viscous forces to the motor,
perturbing its intended function. Because in such enviroments there is no solid
stationary interface nearby for the motor to attach, the motor molecule moves as
a whole and it becomes harder to control the relative motion of its components
and to produce net useful work. By firmly attaching the whole motor to a solid
or a surface, the translational and rotational degrees of freedom associated with
the bulk motion of the motor are suppressed and the positional displacement of
submolecular components becomes the source for generating useful work. On the
other hand, motors buried inside solids or attached to grids and surfaces are at
risk for high dissipation due to coupling between the external drive and different
modes of the solid or substrate.
Kelly et al. [4] reported in 1999 a first example of a synthetic chemically driven
rotary molecular motor capable of performing unidirectional 120◦
rotation. How-
ever, because the sequence of reactions that leads to the initial 120◦
rotation is not
repeatable, the motor did not satisfy one of the basic requirements for a rotary
motor: achievement of repetitive motion. Another example of molecular machines
that can be manipulated by chemical inputs is the family of rotaxane molecules.
They consist of a dumbbell-shaped molecule interlocked with a ring that can travel
along it. Because of their architecture, rotaxanes have potential for applications
in molecular electronics [5], as switching devices [6], or as molecular shuttles [7].
Feringa used light to produce electronic excitation and to induce unidirectional
rotation in a motor that had a high ground-state barrier against rotation [8,9]. The
Feringa motor is much slower [10] in comparison to the rotation speeds displayed
by motor proteins (for example, the rotary bacterial flagellar motor rotates at
speeds of over 100 Hz). Light-driven reversible nanoswitches based on azobenzene
molecules, with potential applications in molecular electronics, as artificial muscles
or molecular motors, have been studied by the Paul Weiss lab [11].](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-17-320.jpg)
![3
Electric fields from scanning tunneling microscope (STM) tips or generated
by applying voltage between nanoelectrodes, have also been used to induce con-
trolled mechanical-like motion in molecules. Coupling to the external drive is
commonly achieved by designing molecules with built-in dipole moments. For
example, electric-field-controlled conformational switches have been constructed
to function as switchable molecular junctions for use in molecular electronics [12].
We discuss a few theoretical and experimental studies of synthetic molecular rotors
chemically attached to surfaces and driven by external electric fields in the intro-
ductory part of Chapter 2. Scanning tunneling microscope tips can also be used
for mechanical manipulation of molecular structures on surfaces. One remarkable
example is the world’s first molecular car [13], which achieved translational motion
and pivoting across a gold surface under the influence of a STM tip. Also, control
of the molecular orientation of double-decker complexes comprised of rare-earth
metals sandwiched between planar ligants on graphite surfaces has been demon-
strated at Penn State [14]. These double-decker complexes are intended for future
applications as molecular rotors attached to surfaces that can be driven by STM
tips, rotating electric fields, or molecular recognition.
Crystalline molecular machines represent another branch of molecular ma-
chines with important applications in nanotechnology. The crystals are built using
molecules that are structurally programmed to respond collectively to external
mechanic, electric, magnetic, or photonic stimuli, in order to fulfill specific func-
tions. M. Garcia-Garibay has studied the dynamics of several different molecular
rotors embedded in carefully engineered molecular crystals and reported rotational
frequencies that range from a few hertz to the gigahertz regime at room tempera-
ture [15]. Possible technological applications target molecular compasses and gyro-
scopes with built-in dipole moments [16] and amphidynamic crystals (i.e., crystals
consisting of solid, rigid frames and highly mobile elements attached to them, such
that the structure displays both phase order and mobility at the same time) [17].
Beams of noble gases have been used by Vacek et al. [18] in a molecular dynam-
ics simulation study, to drive molecular propellers at rotational frequencies of up
to 20-40 GHz. The excitement of rotational motion through momentum transfer
from the gas atoms competed with the induction of pendulum-like motion of the
shaft of the rotary motor, suggesting the need for designing more rigid shafts.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-18-320.jpg)

![5
pends explicitly on the coordinates of the electrons while the coordinates of the
nuclei enter only parametrically, and E is the energy. Also, ˆT, ˆVee and ˆVNe are
the operators describing the kinetic energy of the electrons, the electron-electron
interaction energy and the electron-nuclei interaction energy terms, respectively.
Since the equation still proves too complex for any practical use, a series of ad-
ditional approximations for the energy terms in the electronic Hamiltonian above
are necessary in order to solve it. Based on the choice of the approximation, sev-
eral different methods have been developed, such as ab-initio, semiempirical or
molecular mechanics.
In 1964, a revolutionary approach was developed by Hohenberg, Kohn, and
Sham. Instead of attempting to solve for the many-body wave function of the
Schr¨odinger Eqn. (1.1) in order to find information about the system, they pro-
posed that the complete description of the system can be provided via the ground
state charge density, ρ0(r). This approach results in a dramatic simplification of
the problem by transitioning from the need to know 3N degrees of freedom to solve
for the electronic wavefunction, corresponding to the positions of the N electrons
in the system, to just 3 degrees of freedom for the ground state charge density,
ρ0(r).
Density functional theory (DFT) is based on two theorems of Hohenberg and
Kohn [19]. The first one states that the electron density uniquely determines the
Hamilton operator and thus all properties of the system: ‘the external potential
V ext
(r) is (to within a constant) a unique functional of ρ0; since, in turn V ext
(r)
fixes ˆH, we see that the full many particle ground state is a unique functional of
ρ0’. For our case, V ext
(r) is the potential created by the stationary nuclei, ˆVNe in
Eqn. (1.1). The total energy can be now written as a functional of the electronic
density as follows:
E[ρ] = T[ρ] + Eee[ρ] + V ext
(r)ρ(r)dr = FHK[ρ(r)] + V ext
(r)ρ(r)dr. (1.2)
In Eqn. (1.2) above, the energy components have been separated into those
that depend on the specific system, i.e., ENe[ρ] = V ext
(r)ρ(r)dr, the potential
energy due to the nuclei-electron attraction, and those that are not dependent](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-20-320.jpg)
![6
on the number of electrons and nuclei, and the position and charge of the nuclei,
FHK[ρ(r)]. The Hohenberg-Kohn functional, FHK[ρ(r)], contains the electronic
kinetic energy and the electronic Coulomb interaction and is universal by con-
struction. The first Hohenberg-Kohn theorem only claims the existence of a total
energy functional, E[ρ], but it does not provide the means to solve for the ground
state density that delivers the ground state energy.
The second Hohenberg-Kohn theorem states that FHK[ρ(r)], the functional
that delivers the ground state energy of the system, delivers the lowest energy if
and only if the input density is the true ground state density, ρ0. Therefore, for
any given trial density, the true ground state energy, E[ρ0], satisfies the following
relationship: E[ρ0] ≤ E[ρ]. The ground state energy is obtained by minimization
of the energy functional with respect to electron density. In order to have access
to the exact ground state density and energy, one would need to know the form of
the Hohenberg-Kohn functional, FHK[ρ(r)].
There is a restriction for possible densities to be eligible in the variational
procedure of the second Hohenberg-Kohn theorem: there must exist an external
potential associated with the density of choice, since the energy functional E[ρ] is
only defined for ground state densities for which such an external potential exists.
This is known as the V -representability problem, and many possible trial densities
are known not to be V -representable.
The problem of V -representability for eligible densities can be solved using the
Levy constrained formalism [20], in which only so called N-representability of the
trial densities is required. According to the Levy constrained formalism, a trial
density is N-representable if it satisfies the following conditions: is a non-negative
and finite function, ρ(r) ≥ 0, and it integrates to the total number of electrons,
ρ(r)dr = N.
1.2.2 Kohn-Sham equations
The Hohenberg-Kohn theorems presented in subsection 1.2.1 do not provide any
guidance as to how to construct the Hohenberg-Kohn functional, FHK[ρ(r)].
The breakthrough idea that Kohn and Sham [21] proposed was to replace the
system of interacting electrons with one of fictitious, non-interacting particles that](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-21-320.jpg)
![7
has the ground state density of the original one. This way, the many-body wave-
function of the system could be expressed by an antisymmetrized product of N
one-electron wavefunctions, also known as a Slater determinant, ΘS:
ΘS =
1
√
N!
φ1(r1) φ2(r1) ... φN (r1)
φ1(r2) φ2(r2) ... φN (r2)
...
...
...
φ1(rN ) φ2(rN ) ... φN (rN )
The one-particle orbitals, φi(r), are the eigenfunctions of the one-electron
Hamiltonian:
ˆHKS
φi = iφi. (1.3)
The one-electron operator, ˆHKS
, is defined by the following relationship:
ˆHKS
= −
1
2
2
+ Veff(r), (1.4)
with Veff, the effective potential, chosen such that the total density for the system
of fictitious, non-interacting particles equals exactly the ground state density for
the system of real, interacting electrons:
N
i s
|φi(r, s)|2
= ρ0(r), (1.5)
s in the above equation stands for the spin of the electrons.
In light of the new artificial system, the energy functional, F[ρ], that has been
introduced in the previous subsection, can be further partitioned into three parts:
F[ρ] = T0[ρ] + EH[ρ] + Exc[ρ]. (1.6)
T0[ρ] represents the kinetic energy of a non-interacting electron gas:
T0[ρ] = −
1
2
N
i
φi
2
φi . (1.7)
EH[ρ] is the classical electrostatic Coulomb interaction between electrons:](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-22-320.jpg)
![8
EH[ρ] =
1
2
ρ(r)ρ(r )
|r − r |
drdr . (1.8)
Finally, Exc[ρ] is the so-called exchange-correlation energy:
Exc[ρ] ≡ {T[ρ] + Vee[ρ]} − {T0[ρ] + EH[ρ]} . (1.9)
The exchange-correlation energy term, Exc, contains the non-classical effects of
exchange (i.e., electrons of like spin do not move independently from each other)
and correlation and a part of the kinetic energy. The non-interacting kinetic energy,
T0, is not equal to the real kinetic energy of the interacting system, T, even for
the case when the two systems have the same density. The goal of this energy
partition was to separate the energy into terms that can be easily evaluated, T0[ρ]
and EH[ρ], from the ones that cannot, Exc[ρ].
By rewriting the Hohenberg-Kohn expression for the total energy of the inter-
acting system, given by the Eqn. (1.2), using the Kohn-Sham approach described
by the Eqn. (1.6), one obtains:
E[ρ] = T0[ρ] + EH[ρ] + Exc[ρ] + ENe[ρ]
= T0[ρ] +
1
2
ρ(r)ρ(r )
|r − r |
drdr + Exc[ρ] + VNe(r)ρ(r)dr
= −
1
2
N
i
φi
2
φi +
1
2
N
i
N
j
|φi(r)|2 1
|r − r |
|φj(r )|
2
drdr
+Exc[ρ] −
N
i
M
A
ZA
|r − rA|
|φi(r)|2
dr. (1.10)
Because E is expressed as a function of the orbitals, φi, it can be minimized with
respect to them, while keeping the constraint that φi|φj = δij. The equations
obtained further are known as the one-particle Kohn-Sham equations:
−
1
2
2
+
ρ(r )
|r − r |
dr + Vxc(r) −
M
A
ZA
|r − rA|
φi](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-23-320.jpg)
![9
= −
1
2
2
+ Veff (r) φi = iφi (1.11)
By comparing the Kohn-Sham Eqn. (1.11), with the one-particle equations of
the non-interacting auxiliary system, Eqn. (1.3), one obtains the expression for
the Kohn-Sham potential:
Veff (r) =
ρ(r )
|r − r |
dr + Vxc(r) −
M
A
ZA
|r − rA|
. (1.12)
Notice that Veff itself is a function of the electron density. Therefore, the Kohn-
Sham one-electron equations need to be solved iteratively until self-consistency is
achieved. To summarize, by knowing Veff , one can use the one-particle Kohn-
Sham Eqn. (1.11), to determine the orbitals and further, the ground state density,
Eqn. (1.5), and the ground state energy of the electronic system, Eqn. (1.10).
The Kohn-Sham orbitals are the eigenstates of the auxiliary Hamiltonian for
the system of fictitious, non-interacting particles. Thus, they have no physical
meaning; they are not the wavefunctions for the electrons of the real system.
However, they can be used for the qualitative interpretation of the orbitals in a
molecular system or crystal. Also, the eigenvalues associated to the Kohn-Sham
orbitals have no physical meaning, with one exception: the eigenvalue for the
highest occupied molecular orbital equals the negative of the ionization energy.
1.2.3 The search for approximate exchange-correlation
functionals
Up to this point, the Kohn-Sham approach to solving the many-body Schr¨odinger
equation has been exact. Since the form of the exchange-correlation energy func-
tional, Exc[ρ], is unknown, different approximations are made in order to evaluate
it.
Unfortunately, there is no straightforward way in which the exchange-correlation
energy functional can be systematically improved.
For a homogeneous electron gas or an electron gas with slow-varying charge
density, the exchange-correlation energy functional, Exc, can be approximated as:](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-24-320.jpg)
![10
ELDA
xc [ρ(r)] = xc(ρ(r)) ρ(r) dr, (1.13)
with xc the exchange-correlation energy density function. This approximation is
known as the Local Density Approximation (LDA), and it relies on the assumption
that the exchange-correlation energy depends only on the local value of the charge
density. The exchange-correlation energy per electron of a uniform electron gas of
density ρ(r), xc(ρ(r)), can be split into exchange and correlation contributions as
follows:
xc(ρ(r)) = x(ρ(r)) + c(ρ(r)). (1.14)
The exchange contribution, x(ρ(r)), has the following expression:
x(ρ(r)) = −
3
4
3ρ(r)
π
1
3
. (1.15)
For the correlation contribution, c(ρ(r)), several analytical expressions have
been proposed, based on numerical quantum Monte-Carlo simulations [22].
The LDA approximation works well for solid-state systems, but it fails for
most chemical applications, since molecular systems do not satisfy the restriction
of slow-varying electron density. If the spin densities are used as an input to the
energy functional, instead of the total electron density, ρ(r), the approximation is
known as the Local Spin-Density Approximation (LSD).
Better results are obtained if one takes into account not only the electron den-
sity, ρ(r), but also the gradient of the density, ρ(r), which accounts for the non-
homogeneity of the electron density. The exchange-correlation functionals which
include the gradients of the charge density are known as Generalized Gradient
Approximations (GGA). They can be written generically as:
EGGA
xc [ρ(r)] = xc (ρ(r), | ρ(r)|) ρ(r) dr. (1.16)](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-25-320.jpg)
![11
One of the most popular GGA functional is the Becke exchange functional [23]:
EB88
x [ρ(r)] = βy2
1+6βy sinh−1
[y]
(1.17)
where y = | ρ(r)|
ρ(r)4/3 and the empirical parameter β equals 0.0042.
The gradient-corrected correlation functionals have more complicated analyt-
ical forms and, as with the exchange functionals, contain empirical parameters
fitted to reproduce correlation energies of certain atoms. One popular correlation
functional is the LYP functional, proposed by Lee, Yang and Parr in 1988 [24].
Although, in general, the GGA performs better than LDA, the value of a GGA
functional at a specific point in space still depends only on information about
charge density and its gradient at that very point. Therefore, both approximations
presented above neglect the non-local effects that are very important in some
molecular structures where electrons are delocalized, such as aromatic systems.
The hybrid exchange-correlation functionals have proven very successful in ac-
counting for non-local effects. They include pure exchange and correlation func-
tionals determined within the DFT theory, plus another term which corresponds
to a non-local exact exchange functional determined within the ab-initio Hartree-
Fock approximation (HF). The B3LYP functional [25] is the most used hybrid
functional and consists of a combination of the LSD exchange and correlation
functionals, Becke exchange functional, LYP correlation functional and the exact
HF exchange functional.
Hybrid functionals predict molecular geometries substantially better than LDA
and GGA. For example, for organic molecules, bond lengths computed using
B3LYP show an average deviation from experiment of less than 0.01 ˚A and bond
angles are accurate to within a few tenths of a degree.
Also, with B3LYP functional, the errors in the atomization and the ionization en-
ergies are accurate to within 0.1 eV and 0.2 eV, respectively. The errors in dipole
moments calculated using B3LYP are within 0.04 Debye [26].](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-26-320.jpg)

![13
M
d2
R
dt2
= −
dV
dR
(1.20)
In the equation above, V represents a classic, empirical fit to the quantum
potential energy surface, Etotal(R), and M and R are the mass and the position
for a nucleus in the molecular system, respectively.
In standard classical molecular dynamics (CMD) methods, atoms and molecules
move according to forces dictated by intramolecular and intermolecular classic,
empirical potentials and follow classical trajectories governed by Newton’s laws.
Numerical integration of Newton’s equations of motion is performed using time
steps on the order of 1 fs, which is about 10 times smaller than the period of
oscillation of a hydrogen atom in a molecular system.
The empirical fit to the potential energy surface, V , is also called force field.
The force field defines the coordinates used, the mathematical form of the equations
involving the coordinates, and the parameters adjusted in the empirical fit of the
PES. Usually, the force fields employ a combination of internal coordinates to
describe the bond part of the PES (bond distances, bond angles, torsions), and
interatomic distances to describe the non-bonded interactions between atoms, such
as van der Waals and electrostatic interactions.
In the classical approach, the motion of the atoms in a molecular system re-
sembles the one of vibrating balls connected by Hooke’s springs. Many experi-
mental properties, such as vibrational frequencies, molecular structures or barriers
against rotation about molecular bonds, can be reproduced with a classical force
field because the force field is fit to reproduce relevant observables, and most of
the quantum effects are included empirically. However, there are fundamental lim-
itations of a classical approach, such as electronic transitions, electron transport
phenomena or chemical reactions involving proton transfer. The goal of a force
field is to describe entire classes of molecules with reasonable accuracy.
The Universal force field (UFF) classical potential for molecular mechanics and
dynamics [27–31] is designed to cover the full periodic table of elements. The force
field parameters are estimated using general rules, based only on the element,
its hybridization and its connectivity. The functional form of the UFF classical](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-28-320.jpg)
![14
potential is expressed as a sum of valence or bonded interactions and non-bonded
interactions as follows:
V (R) =
b
Kb
2
(b − b0)2
+
θ
Kθ[C0 + C1 cos(θ) + C2 cos(2θ)]
+
Φ
VΦ
2
[1 − cos(nΦ0) cos(nΦ)] +
γ
Kγ[C0 + C1 sin(γ) + C2 cos(2γ)]
+
i j>i
[
Aij
x12
ij
−
Bij
x6
ij
] +
i j>i
QiQj
Xij
. (1.21)
Equation (1.21) gives the potential energy of an arbitrary geometry of a molecule
with respect to its optimized structure (for example, b0 is the equilibrium bond
length, etc.). The first four terms represent the energy terms due to the bonded
interactions: bond stretching, angle bending, dihedral (or torsional) angle and
improper dihedral angle deformations, respectively. The last two energy terms de-
scribe the non-bonded interactions: van der Waals and electric, respectively. n in
the torsional energy term is an integer and reflects the symmetry with respect to
rotation about the bond. Q represent the point charges associated with the nuclei
of the molecular structure and Xij are the distances between non-bonded atoms.
The UFF force field parameters, i.e., the force constants, the bonded equilibrium
parameters, the electric point charges, etc., are generated using simple combina-
tion rules and the atomic parameters obtained by fitting to experimental data or
ab-initio calculations performed on different individual atoms or molecules. For
example, the equilibrium bond lengths are obtained as the sum of the atomic cova-
lent radii plus corrections for bond order and electronegativity. The covalent radii
are obtained by fitting small sets of molecules.
The UFF classical potential has been applied to different classes of molec-
ular structures, such as organic, main group, transition metal inorganic, and
organometallic compounds, and its performance has been evaluated. The best
performance of the UFF classical potential to predict molecular structures and
conformational energy differences has been reported for organic compounds. For
example, the bond lengths errors are within 0.02 ˚A and the bond angles within 3◦
,
compared to experimental results. The UFF force field failed to describe correctly](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-29-320.jpg)

![Chapter 2
Synthetic caltrop-based molecular
motors driven by rotating electric
field
2.1 Introduction
Electric fields can be used as external stimuli in order to induce intramolecular
conformational changes in molecular motors that carry built-in dipole moments.
Motors chemically attached to surfaces, as opposed to either freely floating in gas
and liquid environments or buried inside solids, seem to offer better prospects to
control molecular scale mechanical motion as access to and control of the dipole-
carrying part of the motor becomes more feasible and direct. Generally, the rotary
motors are comprised of a stationary part (i.e., the stator), which gets attached
to surfaces, an axle and a turning part (i.e., the rotor), that couples to the ex-
ternal drive. Several man-made surface-mounted molecular rotary motors have
been already reported that can achieve periodic and unidirectional rotation under
the influence of external rotating electric fields [32–34]. Also, monolayer films of
such dipolar rotary motors have been investigated by means of dielectric spec-
troscopy [35], in order to exploit their potential for displaying collective rotational
behavior and for applications in ferroelectricity and memory devices.
The dynamical behavior of the individual rotary motors cited above was studied](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-31-320.jpg)
![17
via classical molecular dynamics simulations when driven by external electric fields
with magnitudes between 10−2 V
nm
and up to a couple of volts per nanometer, and
driving frequencies between a few gigahertz and up to several hundred gigahertz.
With periods of rotation on the order of just tens or hundreds of picoseconds, these
rotary motors are intended for applications in nanoelectronics and nanofluidics.
Five different regimes of motion were found for mutually non-interacting rotors,
as a function of the average value of the offset angle between the instantaneous
direction of the field and the dipole: synchronous motion, asynchronous motion,
random driven motion, random thermal motion and hindered motion. Whether
the motor performed in one regime or another was determined by the interplay
between four important quantities in the system: the interaction energy between
the applied driving field and the dipole of the rotor (i.e., −P · E), the magnitude
of the barrier against rotation for the bond that allowed intramolecular conforma-
tional changes, the thermal energy and friction. The review article published by
Kottas et al. [36] discusses in great detail the general theory and basic behavior of
artificial rotary motors.
Dissipation is an important issue in designing machines at both macroscopic
and microscopic scales. In order to improve control over molecular-scale mechani-
cal motion, it is important to understand not only the fundamentals of motion of
the molecular motors but also the fundamentals of their energy dissipation. Tra-
ditionally, friction in rotary motor systems is modeled by effective damping terms
that subsume all of the device-nonrelevant degrees of freedom. For example, the
Langevin equation that describes a one-dimensional rotary motor system is:
I
d2
θ
dt2
=
−∂Vnet
∂θ
− η
dθ
dt
+ ξ(T, t) (2.1)
The molecular rotor has only one explicit degree of freedom, θ, which is associ-
ated with its ability to turn through one torsional angle, while the other molecular
degrees of freedom within the rotary motor comprise the thermal bath. In Eqn.
(2.1) above, I represents the moment of inertia of the dipole-carying rotor about
its rotational axis and Vnet is the total potential that the rotor moves in. Also, η is
the friction constant and ξ the stochastic torque representing thermal fluctuations
in the system (T is temperature and t is time). Since the torsional and nontor-
sional modes in a rotary motor system are intrinsically coupled, the driving force](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-32-320.jpg)
![18
Figure 2.1. Synthetic caltrop-based rotary molecular motor is attached to surfaces
and driven by rotational electric fields. The motor contains a rotor on top, a shaft in
the middle and a three-legged base. The colors of the atoms in the structure are as
follows: carbon-dark blue, hydrogen-green, nitrogen-light blue, oxygen-red, silicon-violet
and sulfur-yellow.
designed to turn the dipole-carrying rotor may also excite other modes within the
system. This gives rise to an increase of friction in the rotary motor system.
Some of the challenges encountered in the rotor systems studied so far are the
decrease of the coupling between the dipole and the underlying surface due to
non-bonded interactions, observed in the case of short rotary motors, and real-
ization of structures that minimize the rotational energy dissipation resulted as a
consequence of resonances with other modes in the system.
Our caltrop-based rotary motor in Fig. 2.1 is another example of a surface-
mounted synthetic molecular machine, engineered to rotate unidirectionally under
the control of rotational electric fields that can be applied between nanofabricated
electrodes situated a couple of micrometers apart. It has been synthesized by J.
Tour at Rice University [37] and is an organic molecule with a size of about 2 nm
in all three spatial directions and a built-in dipole moment in the rotor part of the
molecule. The base of the motor is a molecular structure consisting of four phenyl
rings with tetrahedral spatial orientation which are centered on a silicon atom and
is called a caltrop. A detailed description of the motor is presented in the next
section.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-33-320.jpg)

![20
Figure 2.2. Electrostatic potential energy of a 12.5 Debye permanent dipole moment
in an external electric field of 0.5 V
nm as a function of the angle between the field and
the dipole. The red dashed line represents the sinusoidal fit to the electrostatic potential
energy of the dipole in electric field. The black dashed line is the thermal energy at room
temperature.
rotational barrier enough to allow either ballistic motions or thermally induced
asymmetric hopping on a reasonable experimental timescale.
We perform DFT calculations using the B3LYP hybrid functional and the TZV
basis set as implemented in the GAMESS package [38, 39] in order to study the
static properties of the rotary motor. Since electric dipole moments in organic
molecules are essentially local quantities, we separate out the rotor component,
cap the dangling bond with a hydrogen atom, and calculate the charge distribu-
tion. The static electric dipole moment of the rotor is approximately 12.25 Debye,
aligned to within 2.5◦
of the rotor axis as defined by the two nitrogen atoms at its
ends, and can be separated to a good approximation into contributions from the
amine (32%) and NO2 (68%) groups at either end.
The induced moment is much smaller. At an external electric field of 0.5 V
nm
aligned with the static dipole moment (corresponding to ∼500 V across contacts
separated by one micron), the induced moment is about 3 Debye. Since this in-
duced dipole is symmetric under π rotations of the rotor (rather than 2π rotations,](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-35-320.jpg)
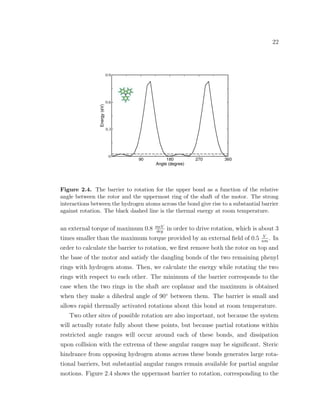
![21
Figure 2.3. Barrier to rotation for the middle bond as a function of the relative angle
between the two benzene rings of the shaft of the motor. The barrier is small and allows
rapid thermally activated rotations about this bond. The black dashed line is the thermal
energy at room temperature.
like the static moment), it does not contribute to the energy difference between
states aligned and anti-aligned to an external electric field. However, it does cause
a small deviation from the sinusoidal dependence of the energy of a rigid static
dipole in an external field, as one can see in Fig. 2.2. The deviation from a curve
corresponding to a rigid dipole, at 90◦
and 270◦
, is due to highly anisotropic po-
larizability for the directions along the rotor and perpendicular to the rotor. The
curve for the electrostatic potential energy of the dipole in the external electric
field of 0.5 V
nm
is symmetric with respect to 180◦
. The rotor can develop a max-
imum torque of about 2.3 meV
deg
or 21 pN · nm. Since the arm of the torque from
the electric field is about 1 nm with respect to the central rotational axis, we ob-
tain that the dipolar unit generates a maximum force of about 20 pN under the
influence of a 0.5 V
nm
electric field. External electric fields of up to 1 - 2 V
nm
can
be achieved experimentally. In comparison, the ATP-synthase biological motor
generates a constant force of 40 pN [2].
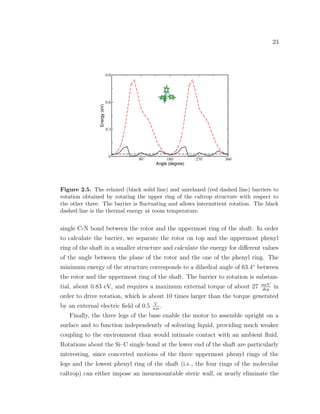
The carbon-carbon triple bond within the shaft is the site of least resistance to
rotation, as one can see in Fig. 2.3. It has a barrier of about 46 meV and requires](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-36-320.jpg)

![25
2.3 Static and dynamic properties of the motor
with classical molecular dynamics
In this section, we study the dynamical behavior of our caltrop-based molecular
motor and possible mechanisms of energy dissipation that can arise from within
the motor structure. We carry out classical molecular dynamics simulations using
the UFF classical potential, as implemented in the TINK molecular dynamics
package [18].
2.3.1 Assessment of quality of UFF classical potential
First of all, we would like to assess how well UFF classical potential describes some
of the static properties of the motor in comparison to DFT. Therefore, we recal-
culate the barriers to rotation for the upper bond, which makes the connection
between the rotor and the shaft, and for the middle bond within the shaft, where
rotation actually occurs. By isolating the degrees of freedom corresponding to up-
per and middle bonds in smaller structures, we change the side functionality, and,
therefore, the charge distribution within these structures, with respect to the one
in the whole motor structure. In order to determine the effect of structure separa-
tion on the barriers to rotation, for the upper and the middle bonds, respectively,
we calculate the barriers to rotation on several different structures using UFF. We
start with the same small structures used for the DFT calculations (see Fig. 2.3
and 2.4, respectively), and keep enlarging the structures, by adding more phenyl
rings. For each of the upper or middle bonds, respectively, we find no variation of
the barrier to rotation with structure size using UFF. Figures 2.6 and 2.7 below
illustrate the comparison between DFT and UFF classical potential for the upper
and middle bonds, respectively.
The barrier to rotation due to the upper bond is very similarly described by
both methods. For a relaxed structure, the dihedral angle between the rotor plane
and the upper ring plane is about 40◦
(20◦
smaller compared to a relaxed structure
in DFT). Barrier calculations using a non-resonant atom-type for the nitrogen
atom at the upper bond between rotor and shaft (i.e., using a set of UFF force
field parameters that neglects the contribution of the π orbitals to the barrier),](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-40-320.jpg)
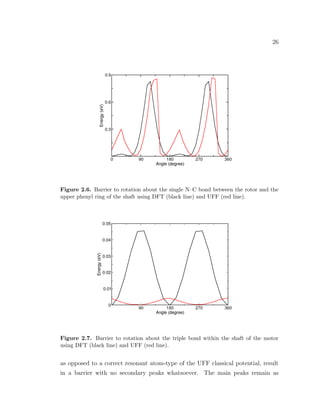
![27
important. This result suggests that, even if expensive DFT barrier calculations
were performed on larger structures, and variations of secondary peaks height with
structure size were obtained, the huge main peaks are independent of the π orbitals
stability. The main peaks are due to the interactions of the hydrogen atoms across
the bond and are almost equal using each method. The character of rotation about
the uppermost bond of the motor is determined by the substantial main peaks,
not the small secondary ones. The small offset between the tips of the main peaks
is just an artifact of the method by which the UFF barrier is calculated. The
secondary peaks in Fig. 2.6, due to the breaking of the π orbital alignment, are
overestimated in UFF compared to DFT. This results in an additional restriction of
the angular intervals available for partial rotations about this bond, which might
overestimate the coupling between the rotational motion of the rotor and high
vibrational modes in the motor.
The middle bond barrier to rotation (see Fig. 2.7) is some 10 times under-
estimated in UFF compared to DFT. Also, notice that the peaks of the barrier
in UFF are shifted 90◦
compared to DFT. Optimization in UFF gives a dihedral
angle of 90◦
between the planes of the two shaft rings for the relaxed structure
of the shaft, while DFT optimizes the shaft with the two rings perfectly aligned.
Other studies for the barrier to rotation, using quantum mechanics calculations, in
a variety of systems containing carbon-carbon triple bonds, and in the absence of
steric hindrance, report extremely close values to the one we found using DFT (see
review article on rotary motors [36]). Although there is a big discrepancy between
the DFT and the UFF description for the barrier to rotation of the middle bond,
the barrier in both cases is very low and comparable to thermal energy, allowing
rapid spontaneous rotation at ambient temperatures. For example, one possible
consequence of this barrier underestimation by UFF classical potential is that the
molecular dynamics simulations might overestimate the upper limit of the field
frequency at which the motor is able to follow the electric field. However, this
effect would become important for weak electric fields and very low temperatures,
when rotation is determined by thermal hopping over the rotational barrier.
The permanent dipole moment of the motor is calculated to be about 15.8 De-
bye using the UFF classical potential and is localized only on the rotor, just as in
DFT. Although it is almost 30% larger compared to the permanent dipole moment](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-42-320.jpg)
![28
of the rotor given by DFT with no external field applied, this value is actually very
close to the one given by DFT for a dipole in an external electric field of about
0.5 V
nm
along the dipole direction. Also, the TINK molecular dynamics package
does not take into consideration any contribution to the dipole due to the pres-
ence of external electric fields. The discrepancy between the values of the dipole
moments as calculated by the two theoretical methods results in overestimation of
the driving force that the electric field applies to the dipole, which further leads
to overestimation of the motor performance at high driving frequencies.
By decomposing the total dipole moment of the rotor (consisting of 50 atoms)
into components parallel and perpendicular to the vertical motor shaft (as defined
by the vector between the Si atom at the base and the middle N atom of the
rotor) we determine that the dipole is oriented 17◦
with respect to the direction
perpendicular to the shaft. We also calculate the relative orientation between
the vertical shaft of the motor and the rotor (as defined by the vector between
the two nitrogen atoms situated at the ends of the rotor) for an optimized motor
structure using UFF classical potential and we find an angle of about 2◦
(NO end
of rotor is lower). Therefore, from a purely geometrical point of view, the rotor
itself is not precisely perpendicular to the shaft. A similar purely geometrical
analysis reveals an angle of about 6◦
between the shaft and the rotor for a motor
structure optimized using an ab-initio level of approximation. We do not know
the direction of the dipole nor the one of the dipole-carrying rotor with respect to
the direction of the shaft in DFT because such a calculation is computationally
expensive. However, since the external rotational electric field lies in the horizontal
plane, it is conceivable that the dipole can induce additional oscillations into the
system, in its attempt to align with the horizontal direction.
2.3.2 Vibrational analysis
We perform vibrational mode frequency calculations on the motor structure in
an attempt to find which soft modes of the motor are likely to couple to the
external electric field and give rise to increased dissipation. We carry out the
vibrational analysis within the approximation of the UFF classical potential using
the partial Hessian vibrational analysis (PHVA) [40] as implemented in the TINK](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-43-320.jpg)
![29
Figure 2.8. Schematic representation of the wobbling modes of the motor with char-
acteristic frequencies of 50 and 80 GHz. The mode corresponds to an oscillation of the
motor shaft with respect to the vertical axis.
molecular dynamic package. The method is extensively used for partially optimized
systems, for example, adsorbates on surfaces [41]. We first perform a constrained
optimization of the motor structure, in which two end atoms from each of the
three legs of the motor are constrained to fixed positions (same atoms are fixed for
all of the MD calculations performed on this motor). Then, we continue with the
partial vibrational mode frequency calculation. The PHVA method diagonalizes
only a subblock of the Hessian matrix to yield vibrational frequencies, the one
corresponding to the non-fixed atoms. We keep those few leg atoms fixed in order
to simulate the attachment of the motor to a surface.
We find that the vibrational spectrum of the motor covers values from 13 GHz
up to a little over 100 THz. Since we drive the motor at frequencies close to the
softer modes of the motor rather than the stiffer ones, and since only the lowest
15% of the vibrational modes are excited at room temperature, we do not consider
the higher end of the vibrational spectrum further. A few soft modes of the motor
have magnitudes within the driving frequency interval for the external electric field
(10-150 GHz) and we present them here in more detail. Two of the modes of the
motor are associated with what we call the shaft wobbling motion (see Fig. 2.8),](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-44-320.jpg)



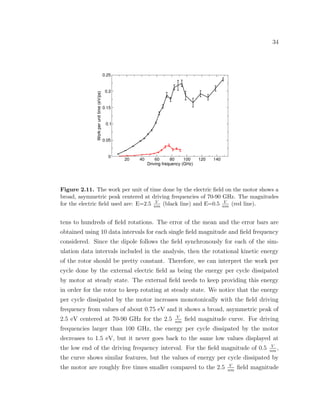

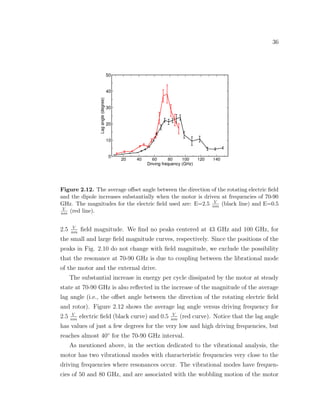
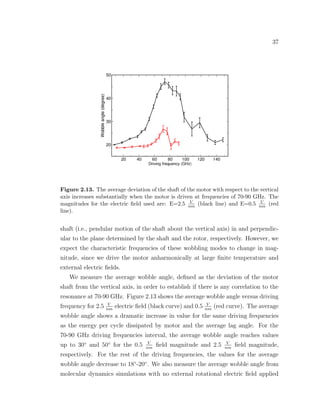

![39
rotational electric field resides. The softness of the shaft is the major design issue
of our rotary motor since it leads to increased dissipation and poor performance
when driven at frequencies close to those of the soft modes of the motor. Another
simulation-based study reported a similar competition between the induction of
rotational motion of the rotor and the excitement of pendulum-like motion of the
shaft for a rotary motor [18]. Therefore, there is a need for designing rotary motors
with more rigid shafts in order to constrain the motion of the rotor to the plane
where the external electric field resides.
2.3.4 Field-free decay analysis
In this section, we examine the field-free decay of the rotational motion. After a
80 ps period of thermal equilibration, we assign additional velocity vectors corre-
sponding to rotational excitations in the range of 10 to 150 GHz to all atoms of
the rotor and we monitor the subsequent motion of the motor. We notice that
the component of the rotor angular momentum along the direction defined by the
shaft of the motor, L(t), decays from its initial value, L(0), rapidly at the begin-
ning and then increasingly slowly. The rotational excitation of the rotor ceases to
be detectable after a maximum of 100 ps. In this time, the rotor transitions from
unidirectional rotation to random rotation.
According to the Langevin Eqn. (2.1) introduced above, a rotor directly cou-
pled to a thermal bath, under the direct influence of a viscous force proportional
to its own speed and that of thermal forces, loses energy and momentum following
an exponential law in time. That is: L(t) = L(0)e− t
τ , where τ represents the decay
time constant or relaxation time. Assuming exponential decay for the component
of the angular momentum of the rotor about the direction defined by the shaft of
the motor, we plot log (L(t)
L(0)
) versus time and fit the data with a straight time in
the attempt to extract a decay time constant for the motor.
We are unable to extract a decay time constant for rotational excitations of the
rotor corresponding to frequencies comparable to the thermal rotation frequency
at room temperature, i.e., 25 GHz. For rotational excitations corresponding to
higher frequencies, the data fit a straight line. We use the inverses of the slopes of
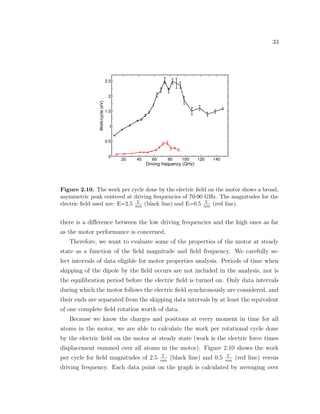
these fits to obtain an average decay time constant for the motor.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-54-320.jpg)
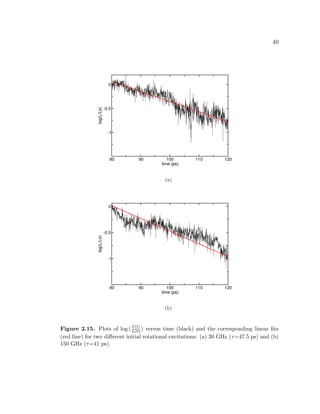
![41
Figure 2.16. The molecular caltrop consists of four identical phenyl rings connected to
a central silicon atom. The structure might function as a molecular gear if the rotary
power generated by mechanically driving one ring would get transmitted to the other
rings via concerted rotations.
Figure 2.15 shows the plots of log (L(t)
L(0)
) versus time (black) and the correspond-
ing linear fits (red line) for two different initial rotational excitations: (a) 30 GHz
(τ=47.5 ps) and (b) 150 GHz (τ=41 ps).
We are not able to detect any significant differencies between the values for
the decay time constants obtained as a function of the frequency. Therefore, we
average over all the runs and obtain a decay time constant of circa 38 ps. In
comparison, different molecular dynamics studies of field-driven surface-attached
rotary motors reported average decay time constants of 83 ps [32] and just a few
picoseconds [33].
The motor thermalizes rotational excitations corresponding to energies of sev-
eral tenths of electron volts in excess of the thermal energy in just several tens of
picoseconds, or the equivalent of up to 10 complete rotations of the rotor. This fast
relaxation is due to a combination between the effect of the thermal fluctuations
within the motor and the coupling between the torsional mode that alows rotation
and the other vibrational modes of the motor.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-56-320.jpg)

![43
Figure 2.17. The four mutually interacting phenyl rings within the molecular caltrop
can be viewed as a sum of six different pairs of two mutually interacting phenyl rings.
For each of these curves, the energy of the two-ring structure is calculated by
allowing only electron relaxation, the nuclei do not relax (i.e., called single point
energy calculations). We find that the highest energy configurations of the two-
ring structure correspond to small values of the two dihedral angles, when the
interactions between the hydrogen atoms coming from the two rings are strong.
The family of relaxed curves, obtained by allowing positional relaxation for both
the electrons and the nuclei (i.e., called optimization calculations), while keeping
the two dihedral angles of the two-ring structure at fixed values, are not shown.
We fit the DFT data for the unrelaxed and/or the relaxed two-ring structures
to a basis set of sine and cosine functions in order to obtain an analytical function
to describe the energy of the two-ring structure as a function of the dihedral angles.
We find that each of the curves in Fig. 2.18 can be fitted well with the following
function:
n
i=0
(A1 sin [iθ1] + B1 cos [iθ1]), with n a positive even integer and A1, B1
real numbers. Since the two phenyl rings and their spatial orientations are iden-
tical, we propose an energy function for the two-ring structure that is symmetric
with respect to the two dihedral angles:
g(θ1, θ2) =
n
i=0
(A1 sin [iθ1] + B1 cos [iθ1])
n
j=0
(A2 sin [jθ2] + B2 cos [jθ2]) ,(2.2)
with i, j and n positive integers and even numbers. The coefficients in front of](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-58-320.jpg)
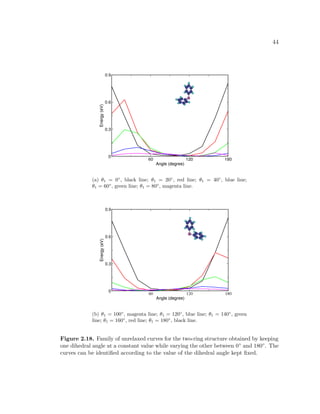

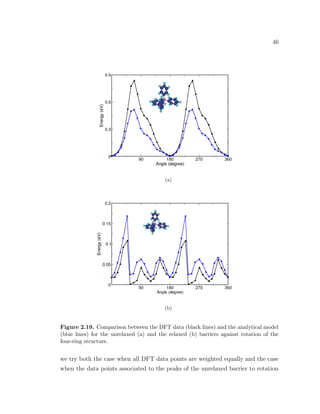

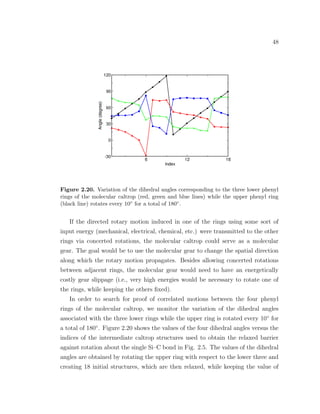
![49
the dihedral angle for the upper ring fixed. Also, the unrelaxed barrier against
rotation in Fig. 2.5 serves as the gear slippage curve.
Figure 2.20 shows clear evidence that the rotation of the upper ring causes the
lower rings to rotate partially with respect to their own rotational axes, but it does
not provide enough information for the range of induced partial rotations or if total
concerted rotations are possible. Additional evaluations of the four dihedral angles
would be necessary on a series of caltrop structures where the next structure in the
series is obtained by alternately rotating the upper ring and relaxing the structure
at the specified angle, and so on. Also, molecular dynamics calculations would
test the performance of the gear at finite temperatures. A comprehensive review
of molecular gears is presented by Kottas et al. [36].
2.5 Conclusions
Our theoretical calculations and simulations using DFT and UFF classical poten-
tial prove that external rotating electric fields with magnitudes accessible experi-
mentally induce unidirectional and repetitive rotation of the dipole-carrying rotor
of the motor. The rotation occurs about the triple bond within the shaft of the
motor. Resonances between the external drive and the soft modes associated with
the deviation of the shaft of the motor with respect to the vertical axis give rise
to a dramatic increase in friction within the motor. This further leads to a lack
of control over the dipole-carrying rotor, designed to move in the horizontal plane
where the external electric field resides.
The molecular caltrop, which makes up the basis of our synthetic rotary motor,
can be described using only four degrees of freedom and may have potential to
function as a molecular machine on its own.
Theoretical investigations can provide guidance to help design motor structures
that allow a higher degree of external control, such that the induced motion is
constrained to a very small number of degrees of freedom.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-64-320.jpg)
![Chapter 3
Power law dissipation in motors
indirectly coupled to a thermal bath
3.1 Introduction
In this section, we present a novel mechanism of dissipation in nanoscale and
molecular-scale motors. We describe a regime in which the deceleration of an
unpowered motor follows a universal power law, rather than a standard exponential
decay.
In larger systems and in traditional treatments of small systems, the motor is
directly and continuously coupled to a large number of degrees of freedom, coming
from the motor’s stator or the environment, which are integrated out into a thermal
bath. The motor is coupled directly to this bath via phenomenological terms such
as viscous damping or Langevin forces [32, 33, 36]. If, for example, the viscous
damping force that acts on the motor is proportional to the speed of the motor,
then the motor dissipates energy and momentum in time following an exponential
law [32,33]. Also, the system has an intrinsic time scale.
As the size of the motors decreases and the design of their structures becomes
highly controllable, it becomes feasible to restrict the coupling between the motor
and the thermal bath to just a very small number of degrees of freedom and to
introduce dissipation in a controlled way.
We study a novel situation where one degree of freedom is pulled out from](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-65-320.jpg)










![61
Figure 3.6. The contour plots show the numbers of elastic collisions in the power law
regime for α = 125, φ = 5◦, 0 < ξ < 2 and 250 < L
σ < 104. Notice that the largest
number of collisions is obtained for very large values of L
σ and ξ close to 1.
3.5 Physical systems
The features of the model that we developed in this section provide guidance for
the design criteria necessary in crafting eligible atomistic structures for a motor
that might show a power law decay of its momentum and energy in time. An
example of a class of physical systems with geometry and properties compatible to
the set up and the assumptions of our model is the class of double-walled carbon
nanotubes (DWCN).
Carbon nanotubes, in general, have already shown potential to function as
nanomotors [42–44] or oscillators [45, 46]. Over the years, experimentalists have
successfully controlled the lengths and the diameters of these systems. They can
reach lengths of milimeters, or even centimeters, and diameters of tens of nanome-
ters. In particular, incommensurate DWCN exhibit the property of superlubricity
(i.e., low friction) between their walls [47,48]. In this case, the potential corruga-
tion between the concentric walls of the nanotube is small (less than 1 meV/atom)
and the tubes can slide easily along each other. As far as their dynamics are](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-76-320.jpg)
![62
concerned, several studies show that the tubes can oscillate relative to each other,
along their longitudinal axes, at frequencies in the gigahertz range [46,49].
Figure 3.7. The outer tube (i.e., the motor) oscillates along its axis relative to the inner
tube and loses momentum via linear and periodic collisions with a small damper. The
damper is directly coupled to the thermal bath (i. e., the inner tube), where it dissipates
the energy gained from the motor.
Since the friction between the walls of the carbon tubes can be negligible, this
may allow the introduction of additional local and well-controlled dissipation into
the system. For example, we can functionalize the inner tube (i.e., the stator) with
a small molecule or even an atom, which would play the role of the damper in the
analytical model (see Fig. 3.7). The oscillating outer tube (i.e., the motor) can
give up momentum or energy via linear and periodic collisions with the damper.
The inner tube would also play the role of the thermal bath where the damper
dissipates the energy gained after the collision with the outer tube. Also, the mass
of the motor could be a few orders of magnitude larger than that of the damper.
We did not carry out any molecular dynamics simulations on atomistic DWCN
structures in this study.
3.6 Conclusions
Most macroscopic motors are immersed in a continuum dissipative fluidic back-
ground, whereas isolated molecular-scale systems are essentially non-dissipative,
since all configurational degrees of freedom are explicit. The transition between](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-77-320.jpg)
![63
these two regimes is unclear, and may take several distinct paths. For example,
if all background degrees of freedom are roughly equally important, then a transi-
tion from an implicit to an explicit dynamics may be best handled by introducing
fluctuation effects onto the continuum to account for the discrete nature of small
systems. However, if the underlying degrees of freedom within the continuum back-
ground occupy a hierarchy of importance, then an alternative means of handling
the transition presents itself: to take successive degrees of freedom out of the con-
tinuum background and into explicit equation of motion one at a time, beginning
from the most important such degree of freedom. Certain molecular motor sys-
tems may satisfy this criterion, if their structure is such that successive collisions
of the main motor degree of freedom with a well-defined substructure dominate
the environmental coupling.
We investigate a situation in which one degree of freedom is pulled out from
the thermal bath and given an explicit equation of motion, interposed between
the bath and the motor. We describe a regime in which the deceleration of an
unpowered motor, coupled to a thermal bath via an explicit degree of freedom,
follows a power law in time with universal exponent of t equal to -1, rather than a
standard exponential decay. We find that the span of the power law regime depends
only on four dimensionless parameters and it can cover up to a few hundred elastic
collisions between the motor and the damper.
Many natural complex phenomena, from real earthquakes, sand piles, and bio-
logical extinctions to stock market fluctuations and traffic jams, follow power laws
in time [50]. However, there is an important distinction between the mechanism of
power law decay that we see in our motor system, and mechanisms of power law
in other systems in nature. Here, a system with no intrinsic time scale displays a
power law decay in time. In other systems, power law time dependence character-
izes multi-time scale processes, such as intramolecular vibrational dephasing [51]
and neuronal response adaptation to stimuli [52].
In the future, it would be interesting to craft and to study real atomistic struc-
tures, guided by the features of the analytical model, to develop real molecular
motors for which energy and momentum decay following a power law in time.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-78-320.jpg)
![Chapter 4
1-Adamantanethiolate monolayer
displacement kinetics follow a
universal form
4.1 Introduction
Self-assembled monolayers (SAMs) are surfaces self-limited to a single, and often
well-ordered in-plane, layer of molecules on a substrate [53]. They are prepared
by adding the solution of the desired molecules onto the substrate, where they
spontaneously self-organize, and washing off the excess. A variety of SAMs can be
formed, using different molecules and substrates. Alkane or cycloalkane molecules
functionalized with thiol head groups (i.e., -SH groups) on gold substrates are a
common example, due to the affinity of sulfur for gold. In these SAMs, the thiol
head groups bind strongly to the substrate, while the molecules pack together
tightly due to the van der Waals forces, but with enough mobility to anneal and
to order.
The patterning and functionalization of surfaces with self-assembled monolay-
ers facilitate the creation of complex well-ordered structures for biosensors [54],
biomimetics [55, 56], molecular electronics [57, 58] or lithography [59–62]. How-
ever, surface diffusion and contamination can hinder the creation of high-quality
structures, especially for lithographic techniques that require multiple deposition](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-79-320.jpg)
![65
steps. Protective layers can assist in controlling deposition, if they can be easily
removed when desired, but otherwise remain impermeable during the fabrication
of surface-bound nanoscale assemblies.
Experimentalists in Paul Weiss’s laboratory showed that 1-adamantanethiolate
(AD) SAMs are labile and can be displaced by short-chain n-alkanethiolates [63].
Although such displacement or exchange reactions are not unique to AD SAMs,
the complete and rapid displacement of one SAM by another under gentle thermal
conditions (room temperature) and dilute concentrations (mM) is unusual [64–66].
The labile nature of AD SAMs makes possible micro-displacement printing, a
technique similar to micro-contact printing, but wherein the patterned molecules
displace an existing SAM in only stamped regions, and the remaining SAM acts as
both a place-holder and a diffusion barrier [60,61]. These diffusion barriers not only
create sharper, higher quality patterns, but also extend the library of patternable
molecules to those otherwise too mobile to retain surface patterns. Despite the
recent interest in and multiple applications of AD SAMs, little is known about
the kinetics of AD SAM displacement. Understanding the displacement kinetics is
important both to achieve higher quality, reproducible chemically patterned films,
and to guide the design of new molecules for use as selectively labile monolayers.
With the help of spectroscopic methods, Paul Weiss’s lab showed that AD
and n-alkanethiolate SAMs have similar sulfur chemical environments [67], so the
displacement is not due to differences in Au–S bond strengths. Also, with the
help of scanning tunneling microscopy (STM), they found that n-alkanethiolate
SAMs are 1.8 times denser than AD SAMs [68]. They concluded that the complete
displacement of the AD SAMs is due to this density difference aided by differences
in van der Waals forces, which provide a substantial thermodynamic driving force.
They estimated that, based only on the energy of breaking/forming a S–Au bond,
the replacement of an AD SAM with an ALK SAM results in a gain of about
35.2 Kcal/mole of AD displaced. Also, using the measured interaction energy of
22.37 Kcal/mole for a C12 SAM, the replacement of an AD SAM with an ALK
SAM would result in a intermolecular interaction energy gain between 17.9 and
40.3 Kcal/mole of AD replaced.
Imaging with STM revealed that displacement begins with a rapid nucleation
phase, where n-dodecanethiolate (C12) molecules insert at defect sites of the AD](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-80-320.jpg)
![66
Figure 4.1. A) Schematic representation of the displacement of AD molecules by
C12 molecules. n-dodecanethiolate (C12) molecules insert at defect sites of the AD
SAM during a rapid nucleation phase. B) STM image of the real process: the (C12)
islands grow radially into domains that coalesce and eventually fully displace the original
monolayer.
SAM, followed by radial growth into domains that coalesce and eventually fully
displace the original monolayer [69]. The defects consist of both randomly dis-
tributed single-atom-deep vacancy islands in the gold substrate (from lifting the
Au{111} surface reconstruction during self-assembly) [70–72] and rotational/tilt
domain boundaries in the original SAM. Figure 4.1 shows a schematic representa-
tion of the displacement of the AD molecules by the C12 molecules and an STM
image of the real process.
In order to study the quantitative kinetics of the solution-phase displacement of
AD SAMs by C12 on Au{111}, they used Fourier transform infrared spectrometry
(FTIR). Below, we present briefly the experimental results of our collaborators.
For a more detailed description of the experimental methods and results, we direct
the reader to the article we published together with our collaborators [73].
4.2 Experimental results
The experimental procedure comprises several steps. First, the experimentalists
fabricate the AD SAMs by immersing flame-annealed Au{111} on mica substrates
into ethanolic solutions of 1-Adamantanethiol molecules with concentrations of
10 mM. After 24 hr deposition from solution, the gold substrates are removed,
rinsed with ethanol, and dried under a stream of nitrogen. The newly created
AD SAMs are investigated immediately after preparation, in order to asses their](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-81-320.jpg)
![67
0.0000
0.0005
0.0010
0.0015
0.0020
2800 2850 2900 2950 3000
2850
2877
2911
2919
2935
2934
2963
2850
1-adamantanethiolate
n-dodecanethiolate
Wavenumber (cm-1)
Absorbance(a.u.)
Figure 4.2. Infrared spectra of the C-H stretch region of a AD SAM (black) and a
C12 SAM (grey), showing their spectral overlap.
quality. Preliminary FTIR spectra are acquired to verify the absence of impurity-
related features and the presence of the CH2 stretch at 2911 ± 1 cm−1
, both
indicative of a well-ordered AD SAM [69,74].
Next, they place the well-ordered AD SAMs in ethanolic C12 solutions of
specified concentration in order to achieve displacement. Every six minutes, the
SAMs are removed from solution, rinsed with ethanol and dried with nitrogen, a
FTIR spectrum is recorded, and the sample is returned to C12 solution for the
next incremental exposure. Displacements are no longer incrementally monitored
once the signals plateau; instead, the samples are placed in C12 solution overnight
to allow slow reordering and annealing and thereby achieve saturation coverage.
They obtain the infrared spectra of the adsorbed species on substrates from
400 to 4000 cm−1
. The region from 2800 to 3000 cm−1
contains the CH2 and CH3
stretch modes of the aliphatic and carbon-cage tails of the thiolated molecules.
Figure 4.2 shows the typical spectra of an AD SAM (black) and a C12 SAM
(grey), both recorded after 24 hr deposition. Several absorption peaks overlap in
this region: CH2 symmetric stretches (2850 cm−1
for both AD and C12 SAMs)
and CH2 asymmetric stretches (2911 cm−1
for AD SAMs and 2919 cm−1
for C12
SAMs) [74]. The peaks that do not overlap correspond to the CH3 symmetric and
asymmetric stretching modes of the C12 SAM, at 2877 and 2963 cm−1
, respectively
[75].](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-82-320.jpg)
![68
0.000
0.005
0.010
0.015
0.020
0.025
0.030
0.035
2850 2900 2950 3000 30502800
Absorbance(a.u.)
Wavenumber (cm-1)
0 20 40 60 80 100
0.000
0.001
0.002
0.003
0.004
0.005
0.006
0.007
IntegratedAbsorbance(a.u.)
Exposure Time (min)
0 min
18 min
36 min
54 min
A B
Figure 4.3. A) Representative FTIR spectra of a 0.11 mM C12 solution displacing
an AD SAM. The 2877 cm−1 peak, corresponding to the CH3 symmetric mode, is
highlighted. B) A kinetic curve derived from the FTIR spectra by plotting the integrated
C12 CH3 symmetric mode peak versus deposition exposure time. The open squares
represent the integrated absorbance for each of the four spectra shown on the left.
These spectra provide peak intensities and peak positions as a function of im-
mersion time and solution concentration for two experimental trials per concentra-
tion. Figure 4.3A displays four spectra obtained at increasing immersion times in
a 0.11 mM C12 solution; each is a sum of contributions from both AD and C12.
Figure 4.3B plots the integrated 2877 cm−1
peak intensity as a function of exposure
time. Molecular orientation and lattice crystallinity affect the spectrum; for exam-
ple, shifts in the CH2 asymmetric mode track monolayer order and crystallinity.
However, the symmetric and asymmetric CH3 stretching modes are not sensitive
to the orientation of the C12 molecule, because of the tetrahedral coordination of
the three relevant hydrogens. Therefore, the strength of the symmetric CH3 mode
directly measures the C12 surface coverage [75]. As one can see in Fig. 4.3A, this
mode is initially very weak. After about 12 minutes of exposure to 0.11 mM C12,
the intensity of the 2877 cm−1
peak increases rapidly and eventually dominates
the spectrum. After 28 minutes, no signal from AD molecules can be detected
by FTIR and the spectrum is nearly identical to that of a pure C12 SAM. Dis-
placement becomes very slow at around 92% of the final saturation intensity and
approaches final saturation only after 24 hrs in solution. A similar but faster time
evolution is observed at higher displacement solution concentrations.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-83-320.jpg)
![69
4.3 Modelling of the kinetics of the displacement
process
In this section, we present the modelling of the kinetics of the displacement pro-
cess. Several experimental techniques [76–78] established that the growth rates
of alkanethiolate monolayers on bare Au{111} surfaces obey Langmuir kinetics
law, where the growth rates of adsorption are proportional to the number of un-
occupied adsorption sites on the surface. In our attempt to model the C12/AD
displacement kinetics, we consider several variants of the Langmuir model as eligi-
ble kinetics models, and in addition, a purely diffusion-controlled adsorption model
and two models based on island growth.
The simplest case, first-order Langmuir kinetics, is based on several assump-
tions: 1) all surface adsorption sites are equivalent; 2) a surface site is filled by
reaction with one molecule; molecules cannot adsorb in the regions around a site,
nor can multilayers form; 3) the number of sites remains unchanged during the re-
action; 4) there are no lateral molecular interactions, no interactions between the
adsorbing molecules and the pre-adsorbed ones nor the solvent molecules; and 5)
temperature is constant. The consequence of some of these assumptions is that the
adsorption rate is uniform across the entire available surface. By integrating the
relationship between the adsorption rate and the number of unoccupied adsorption
sites on the bare surface, dθ
dt
= κ(1 − θ), one obtains [79]:
θ(t) = 1 − e−κt
, (4.1)
where θ is the time-dependent surface coverage and κ is the rate constant.
Despite its simplicity, first-order Langmuir kinetics describes monolayer uptake
curves on bare gold surfaces fairly well. It has also been used to model the molec-
ular exchange of n-octadecanethiolate SAMs by radiolabeled n-octadecanethiol
molecules, although the reaction took 50 hrs and reached only ∼60% comple-
tion [66]. If the onset of surface coverage growth is delayed, then a time offset can
be introduced into the Langmuir equation above: θ(t) = 1 − e−κ(t−tc)
[80,81].
First-order Langmuir kinetics has been extended to account for diffusion-limited
kinetics [82], second-order processes [64, 82] and intermolecular interactions [83].](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-84-320.jpg)
![70
When growth is limited by the diffusion of the molecules from the bulk solution to
the surface, one obtains the square-root time dependence associated with molecular
diffusive random walks:
θ(t) = 1 − e−
√
κt
. (4.2)
The diffusion-limited Langmuir model has been used successfully to describe the
adsorption kinetics of alkanethiol molecules on bare gold surfaces from very dilute
solutions ( 100 µM) [84].
If the rate of adsorption is second-order in the thiol concentration, the analytical
expression for the growth of the surface coverage over time becomes:
θ(t) = 1 −
1
1 + κt
. (4.3)
Second-order Langmuir adsorption kinetics has been used to describe ligand-exchange
reactions on ligand-stabilized nanoparticles. The rate of the reaction was taken to
depend on the concentration of the exchanging thiol both in solution and on the
surface [64].
For the purely diffusion-controlled adsorption, the time-dependent coverage
follows from the solution of the diffusion equations for a semi-infinite medium
[66,85]:
θ(t) =
√
κDt, (4.4)
where
κD =
4D
πB2
ml
, (4.5)
D is the diffusion constant and Bml is the number of molecules per unit area in the
full monolayer. The rate of adsorption is then proportional to the flux of molecules
to the surface. Since this model does not account for saturation or the depletion
of adsorption sites, it could only apply to the initial stages of growth.
The kinetics of phase transformations that occur via nucleation and island
growth were first modeled by Johnson, Mehl, Avrami and Kolmogorov (JMAK) in
the 1940s to describe metal alloy phase transformations [86–89].](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-85-320.jpg)
![71
The JMAK model was originally derived in the framework of the following ex-
perimentally supported assumptions: 1). the nuclei are distributed randomly and
uniformly throughout the volume or the surface of the system wherein the phase
transformation occurs; 2). the growth rates of the newly formed grains/islands are
constant in time and uniform in all spatial directions; 3). growth ceases wherever
adjacent grains/islands impinge on each other; 4). the volume within which the
phase transformation occurs is infinite, there are no effects associated with finite
boundaries. The second assumption is equivalent to the assumption that the rate
at which the total transformed volume or surface grow is proportional to the total
surface area of the grains or the total perimeter of the islands, respectively.
The total number of nuclei distributed randomly and uniformly throughout
the volume/surface of the old phase changes as the phase transformation pro-
ceeds. Some of them start growing while others get swallowed up by the growing
islands of the new phase. Avrami finds that the time-dependent total transformed
volume/surface of the newly formed phase, V (t), can be expressed as a functional
of the number of nuclei, N(t), i.e., V (t) = V [N(t)] [86, 87]. Thus, at any time t,
the total transformed volume depends upon the values of N throughout the entire
time interval [0, t]. The functional can be then expanded as a Taylor series of sim-
ple functionals of different orders: linear, quadratic etc. The physical significance
of the various order terms in the series expansion can be extracted if one considers
a system in which the nuclei give rise to islands that grow unimpeded by other
neighboring growing islands. Because the islands overlap, the total transformed
volume obtained by summing over all the islands, called the ’extended’ volume, Ve,
overestimates the actual total transformed volume. Ve represents the linear term
in the series expansion of V [N(t)].
Let us denote V1 to be the transformed volume lying solely in the nonoverlap-
ping regions of Ve, V2 as the transformed volume lying solely in double overlapping
regions and so on. Both V and Ve can be expressed as a function of the volumes
corresponding to n-overlapping regions as follows:
V = V1 + V2 + ... + Vn + ..., (4.6)
Ve = V1 + 2V2 + ... + nVn + ... (4.7)](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-86-320.jpg)
![72
After lengthy calculations, Avrami proves that the time-dependent total trans-
formed volume/surface of the newly formed phase, V (t), can be expressed using of
the total extended transformed volume, Ve, via this relationship: V (t) = 1−e−Ve(t)
.
Next, the extended transformed volume can be easily evaluated by using the rates
of nucleation and growth for the physical system at hand, and is found to have the
following form: Ve(t) = (κt)n
, where κ and n are constants.
The JMAK equation relates the evolution in time of the macroscopic fraction
of the newly formed phase (in our case, the C12 SAM surface coverage), with the
microscopic mechanisms of nucleation and growth:
θ(t) = 1 − e−(κt)n
, (4.8)
where κ is the rate constant of the transformation (in our case, the rate of displace-
ment). The Avrami exponent, n, reflects the dimensionality of the system and the
time dependence of the nucleation: n=2 for a two-dimensional system wherein nu-
cleation proceeds rapidly to completion (site-saturated nucleation, JMAK2), and
n=3 for a two-dimensional system wherein the nucleation rate is constant in time
(constant-rate nucleation, JMAK3). In JMAK3, more nucleation sites become
available as the transformation proceeds, such as in certain glass ceramics [90].
The JMAK model has later been extended to address heterogeneous nucleation
[91], non-uniform island growth rates [92, 93], and boundary constraints [94–96],
and has been applied to describe a variety of physical systems including oxidizing
metal surfaces [97,98], graphite-diamond transformations [99], and the crystalliza-
tion of thin films [100] or proteins [101].
Figure 4.4 shows least-squares fits of several different kinetic models to the
C12 coverage versus exposure time for a 0.01 mM C12 displacement solution.
Notice that of all the kinetic models considered, only JMAK2 (Eqn. (4.8) with
n = 2) fits the data. A similar conclusion applies across the full range of all C12
concentrations studied, from 0.01 to 1.0 mM, as shown in Fig. 4.5.
Although the diffusion-limited models successfully described the growth of
n-alkanethiolate from very dilute solutions onto bare gold surfaces [76], they do
not fit the experimental surface coverage curves for our physical system, where
adsorption occurs on a slower time scale (minutes as opposed to seconds) and the
solution concentrations are much higher (mM as opposed to µM).](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-87-320.jpg)
![73
pure diffusion
first-order Langmuir
first order Langmuir (tc=23 min)
diffusion-limited Langmuir
second-order Langmuir
site-saturated nucleation JMAK2
constant-rate nucleation JMAK3
0 50 100 150 200 250 300 350
0.0000
0.0012
0.0025
0.0037
0.0050
0.0062
Exposure Time (min)
IntegratedAbsorbance(a.u.)
Figure 4.4. A representative 0.01 mM C12 uptake curve with least-squares fits to pure
diffusion (green), first-order Langmuir (purple), first-order Langmuir with an onset of
growth at 23 min (orange), diffusion-limited Langmuir (grey), second-order Langmuir
(black), site-saturated nucleation JMAK2 (red), and constant-rate nucleation JMAK3
(blue) models.
0 20 40 60 80
0.0
0.2
0.4
0.6
0.8
1.0
0.01 mM
0.035mM
0.11 mM
0.33 mM
0.55 mM
0.77 mM
1.00 mM
Exposure Time (min)
θC12
Figure 4.5. n-Dodecanethiolate monolayer formation by the displacement of an AD
SAM as a function of concentration. Solid lines are least-squares fits based on the site-
saturated nucleation model JMAK2 (Eqn. (4.8) with n=2).
The failure of Langmuir-based models, wherein adsorption is equally probable at all
unoccupied sites, is consistent with the STM observations that adsorption begins
at defect sites (i.e., not within the interiors of AD domains) and then proceeds at
the AD-C12 domain boundaries [69]. The rapid nucleation across these preexist-
ing active sites accounts for the success of JMAK2 over JMAK3.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-88-320.jpg)
![74
-2.0 -1.5 -1.0 -0.5 0.0
-2.2
-2.0
-1.8
-1.6
-1.4
-1.2
-1.0
-0.8
Log[κ(min-1)]
Log[ C12 Concentration (mM) ]
log κ = 0.5 log[C12] - 1.0
Figure 4.6. The displacement rate constant versus C12 concentration on a logarithmic
scale. The slope of 0.50 ± 0.05 implies that the rate constant is proportional to the
square-root of the C12 concentration.
Although several elaborations upon JMAK2 (incorporating boundary effects or
non-uniform growth rates) could be derived, such subtle distinctions are not re-
solved by the experimental data.
The fits to JMAK2 also provide a measure of the displacement rate constant κ
for each C12 concentration. Figure 4.6 plots the logarithm of κ for all experimen-
tal trials against the logarithm of the C12 solution concentration. The slope of
0.50 ± 0.05 suggests that the displacement rate is proportional to the square-root
of the C12 solution concentration, assuming that the density of nucleation sites
is roughly constant across all samples. This result is surprising, since insertion
and displacement is expected to be a unimolecular process (a bimolecular process
would yield a slope of two, not one-half). Half-order kinetics can arise, for exam-
ple, when the displacement of Y by X actually proceeds by the decomposition of
a predominant bimolecular state X2 that generates X. In our case, the half-order
kinetics suggests that the thiol ends of two adjacent C12 molecules might stick
together in solution, before displacing the AD molecules on the surface.
In addition to specific conclusions regarding the relative quality of fits to differ-
ent kinetic models, one can also analyze the displacement curves on a model-free
basis, to extract powerful general conclusions regarding the number and character
of any component sub-processes. Consider a general physical process as a compo-](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-89-320.jpg)

![76
constants, but as mentioned above, this scale-free universal behavior is model-
independent. The crystallization of metal alloys [86], the growth of diamond on
deformed silicon surface [102], or the oxidation of nickel surface [97,98] are other
examples of scale-free processes.
4.4 Conclusions
n-Dodecanethiol molecules in solution displace 1-adamantanethiolate self-assembled
monolayers on Au{111}, leading to complete molecular exchange. Fast insertion of
n-dodecanethiolate at defects in the original 1-adamantanethiolate monolayer nu-
cleates an island growth phase, which is followed by an eventual slow ordering of the
n-dodecanethiolate domains into a denser and more crystalline form. Langmuir-
based kinetics, which describe alkanethiolate adsorption on bare Au{111}, fail to
model this displacement reaction. Instead, a Johnson-Mehl-Avrami-Kolmogorov
model of perimeter-dependent island growth yields good agreement with exper-
imental data obtained over a hundred-fold variation in n-dodecanethiol concen-
tration. Rescaling the growth rate at each concentration collapses all the data
onto a single universal curve, suggesting that displacement is a purely geometri-
cal, scale-free process. The rate of displacement varies as the square-root of the
n-dodecanethiol concentration across the 0.01–1.0 mM range studied.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-91-320.jpg)
![Chapter 5
Lanthanide double-decker complexes
as potential rotary motors
5.1 Introduction
Planar organic ligands, such as porphyrin (Pr), phthalocyanine (Pc), and naph-
thalocyanine (Nc) form complexes with metal ions, wherein the metal lies between
two ligands, known as metal double-decker complexes (DD). These structures have
important applications as sensors [103], molecular memory [104], and rotary mo-
tors [105].
X-ray diffraction analysis [106,107] reveals that lanthanide double-decker com-
plexes have sizes of 1-2 nm. Ring-to-ring separations between the internal faces of
the ligands vary between 2 ˚A and 3 ˚A, depending on the type of ligand and metal
ion. Strong π-π interaction between ligands results in staggered DD conformations,
with twist angles of 45◦
between the rings.
Aida and co-workers were the first to control the rate of rotation, to produce a
rotary motor capable of more than one speed [105,108]. They used a bisporphyri-
nate double-decker complex, with a cerium or zirconium ion sandwiched between
the two Pr ligands, that rotate with respect to one another. They found rotational
rates on the order of 10−6
to 10−4
Hz. Also, they discovered that, by reducing the
cerium complex led to the rotation of the Pr ligand being accelerated more than
300-fold.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-92-320.jpg)
![78
Oxidation of the zirconium complex, decelerated the rotation of the Pr ligand by
a factor of 21 or 99, depending on the oxidation state of the complex.
Experimentalists in Paul Weiss’s lab at Penn State investigated the potential
of lanthanide DD complexes to function as rotary motors. They developed and
applied design rules that enabled precise control over adsorption orientation and
spacing of lanthanide DD molecule arrays on substrates. They selectively attached
certain ligands within DD molecules to the graphite surface and oriented the coun-
terpart ligands off the surface [109,110]. Also, they controlled the intermolecular
distances between neighboring DD molecules on surface by varying the size of the
ligands adsorbed to the surface and by coadsorbing DD molecules with single-
ligand molecules [111]. In a DD rotary motor attached to a substrate, the metal
ion plays the role of the shaft of the motor, while the upper and the lower ligands
represent the rotor and the stator, respectively. Scanning tunneling microscopy
studies are yet to observe upper ligand rotation in any of the structures analyzed.
Our goal is to calculate the barriers to rotation for individual DD molecules
investigated by experimentalists, in an attempt to evaluate their potential as rotary
motors.
5.2 Barriers to rotation for lanthanide DD
complexes using DFT
In this section, we present the results for barriers to rotation for several different
DD molecules using DFT. We consider a luthetium metal ion and the following
combinations of ligands with no side-substituents: Pr/Pc (i.e., (Pr)Lu(Pc)), Pr/Nc
(i.e., (Pr)Lu(Nc)), Pc/Pc (i.e., (Pc)Lu(Pc)) and Pc/Nc (i.e., (Pc)Lu(Nc)).
Figures 5.1 and 5.2 show schematic representations of (Pc)Lu(Pc) and (Pr)Lu(Nc)
structures, respectively.
In order to calculate the barriers to rotation, we first optimize the DD molecules.
We find that the minimum energy structures correspond to staggered conforma-
tions with a twist angle of 45◦
for all structures investigated. Next, we calculate
the energies of the eclipsed conformations, which correspond to a skew angle of 0◦
.
The energy barriers to rotation are obtained as the difference in energy between](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-93-320.jpg)

![80
(a) (b)
Figure 5.3. Staggered conformation of optimized (Pr)Lu(Pc) double-decker complex
using DFT. (a) Side view. (b) Top view. Atom colors are as follows: Lu yellow, N blue,
C and H green.
ligand with Nc does not give rise to a significant change in the energy barrier to
rotation. The outer Nc rings are situated too far away from the upper ligand, and
do not contribute to their mutual π-π interactions.
Similarly, the barriers to rotation for (Pc)Lu(Pc)) (see Fig. 5.4) and (Pc)Lu(Nc))
are almost equal, i.e., 0.94 eV. The intraplanar distance between the Pc ligands
in staggered conformation is 2.71 ˚A, calculated as the distance between the planes
determined by the central nitrogen atoms of each ligand. The N–Lu bond lengths
are 2.44 ˚A, with Lu ion at the same distance from each of the two Pc ligands.
The barriers to rotation of these Lu DD complexes are substantial and allow
only thermally activated hopping between adjacent staggered configurations of
the DD structures. By assuming an average value of −20 cal/mol/K [103] for the
activation entropy between the staggered and eclipsed conformations, and using the
Arrhenius formula (i.e., r = ωe
− ∆G
kBT
, with r, the rate of rotation, ω, the attempt
frequency, and ∆G, the activation free energy.), we estimate thermal rotational
rates on the order of 10−8
to 10−5
Hz at room temperature.
As a result of lanthanide contraction, we expect to obtain smaller barriers
to rotation when Lu is replaced by other lanthanide metal ions. However, the
optimization of DD complexes with lanthanide metal ions other than Lu proves
challenging, due to their incomplete 4f shell. In particular, it is rather difficult to
achieve electron density convergence for these systems. For example, the electron](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-95-320.jpg)
![81
(a) (b)
Figure 5.4. Staggered conformation of optimized (Pc)Lu(Pc) double-decker complex
using DFT. (a) Side view. (b) Top view. Atom colors are as follows: Lu yellow, N blue,
C and H green.
density of the system can oscillate between two different electron density values,
whithout ever converging. Such issues are studied by a branch of mathematics
called chaos theory, and several different methods have already been developed to
address convergence difficulties in metal complexes [112].
5.3 Conclusions
Our preliminary results show that, the rotational barriers for a variety of DD
molecules, with no side-substituents attached to the ligands, and a luthetium ion
as shaft, are substantial and allow rotation only via thermally activated hopping.
For the future, it would be interesting to study the variation of rotational barriers
with size of the metal ion and its oxidation state, and size and position of ligand
side-substituents.
Experimentalists plan to continue their investigations of the rotation of DD
molecules with asymmetric rotors, which allow direct observation of the hopping
motion.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-96-320.jpg)
![Bibliography
[1] Noji, H., R. Yasuda, M. Yoshida, and K. Kinosita (1997) “Direct
observation of the rotation of F1 − ATPase,” Nature, 386, p. 299.
[2] Yasuda, R., H. Noji, K. Kinosita, and M. Yoshida (1998) “F1 −
ATPase is a highly efficient molecular motor that rotates with discrete 120◦
steps,” Cell, 93, p. 1117.
[3] Schliwa, M. and G. Woehlke (2003) “Molecular motors,” Nature, 422,
p. 759.
[4] Kelly, T. R., H. D. Silva, and R. A. Silva (1999) “Unidirectional rotary
motion in a molecular system,” Nature, 401, p. 150.
[5] Green, J. E., J. F. Stoddart, and J. R. Heath (2007) “A 160-kilobit
molecular electronic memory patterned at 1011
bits per square centimetre,”
Nature, 445, p. 414.
[6] Bissell, R. A., E. Cordova, A. E. Kaifer, and J. F. Stoddart (1994)
“A chemically and electrochemically switchable molecular shuttle,” Nature,
369, p. 133.
[7] Berna, J. and F. Zerbetto (2005) “Macroscopic transport by synthetic
molecular machines,” Nature Materials, 4, p. 704.
[8] Koumura, N., R. Zijlstra, R. A. van Delden, N. Harada, and
B. Feringa (1999) “Light-driven monodirectional molecular rotor,” Nature,
401, p. 152.
[9] van Delden, R. A., M. ter Wiel, M. Pollard, J. Vicario,
N. Koumura, and B. Feringa (2005) “Unidirectional molecular motor
on a gold surface,” Nature, 437, p. 1337.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-97-320.jpg)
![83
[10] Vicario, J., M. Walko, A. Meetsma, and B. Feringa (2006) “Fine
tuning of the rotary motion by structural modification in light-driven unidi-
rectional molecular motors,” J. Am. Chem. Soc., 128, p. 5127.
[11] Kumar, A. S., T. Ye, T. Takami, B. Yu, A. K. Flatt, J. M. Tour,
and P. S. Weiss (2008) “Reversible photo-switching of single azebenzene
molecules in controlled nanoscale environments,” Nano Letters, 8, p. 1644.
[12] Troisi, A. and M. A. Ratner (2004) “Conformational molecular recti-
fiers,” Nano Letters, 4, p. 591.
[13] Shirai, Y. S., A. J. Osgood, Y. Zhao, K. F. Kelly, and J. M. Tour
(2005) “Directional control in thermally driven single-molecule nanocars,”
Nano Letters, 5, p. 2330.
[14] Takami, T., T. Ye, D. P. Arnold, K. Sugiura, R. Wang, J. Jiang,
and P. S. Weiss (2007) “Controlled adsorption orientation for double-decker
complexes,” J. Phys. Chem. C, 111, p. 2077.
[15] Khuong, T., J. Nunez, C. Godinez, and M. Garcia-Garibay (2006)
“Crystalline molecular machines: A quest toward solid-state dynamics and
function,” Acc. Chem. Res., 39, p. 413.
[16] Dominguez, Z., T. Khuong, H. Dang, C. Sanrame, J. Nunez, and
M. Garcia-Garibay (2003) “Molecular compasses and gyroscopes with
polar rotors: synthesis and characterization of crystalline forms,” J. Am.
Chem. Soc., 125, p. 8827.
[17] Garcia-Garibay, M. (2005) “Crystalline molecular machines: Encoding
supramolecular dynamics into molecular structure,” Proc. Natl. Acad. Sci.,
102, p. 10771.
[18] Vacek, J. and J. Michl (1997) “A molecular ”Tinkertoy” construction kit:
Computer simulation of molecular propellers,” New J. Chem., 21, p. 1259.
[19] Hohenberg, P. and W. Kohn (1964) “Inhomogeneous electron gas,” Phys.
Rev. B, 136, p. 864.
[20] Levy, M. (1982) “Electron densities in search of Hamiltonians,”
Phys. Rev. A, 26, p. 1200.
[21] Kohn, W. and L. J. Sham (1965) “Self consistent equations including
exchange and correlation effects,” Phys. Rev. A, 140, p. 1133.
[22] Ceperley, D. M. and B. J. Alder (1980) “Ground state of the electron
gas by a stochastic method,” Phys. Rev. Lett., 45, p. 566.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-98-320.jpg)
![84
[23] Becke, A. D. (1988) “Density-functional exchange-energy approximation
with correct asymptotic behavior,” Phys. Rev. A, 38, p. 3098.
[24] Lee, C., W. Yang, and R. G. Parr (1988) “Development of the Colle-
Salvetti correlation-energy formula into a functional of the electron density,”
Phys. Rev. B, 37, p. 785.
[25] Stephens, P. J., J. F. Devlin, C. F. Chabalowski, and M. J. Frisch
(1994) “Ab initio calculation of vibrational absorption and circular dichroism
spectra using density functional force fields,” J. Phys. Chem., 98, p. 11623.
[26] Koch, W. and M. C. Holthausen (2001) A chemist’s guide to density
functional theory, John Wiley & Sons, Weinheim.
[27] Rappe, A. K. and W. A. Goddard (1991) “Charge equilibration for
molecular dynamics simulations,” J. Phys. Chem., 95, p. 3358.
[28] Rappe, A. K., C. J. Casewit, K. S. Colwell, W. A. Goddard, and
W. M. Skiff (1992) “UFF, a full periodic table force field for molecular
mechanics and molecular dynamics simulations,” J. Am. Chem. Soc., 114,
p. 10024.
[29] Casewit, C. J., K. S. Colwell, and A. K. Rappe (1992) “Application
of a universal force field to organic molecules,” J. Am. Chem. Soc., 114, p.
10035.
[30] Casewit, C., K. S. Colwell, and A. K. Rappe (1992) “Application of
a universal force field to main group compounds,” J. Am. Chem. Soc., 114,
p. 10046.
[31] Rappe, A. K., K. S. Colwell, and C. J. Casewit (1993) “Application
of a universal force field to metal complexes,” Inorg. Chem., 32, p. 3438.
[32] Vacek, J. and J. Michl (2001) “Molecular dynamics of a grid-mounted
molecular dipolar rotor in a rotating electric field,” Proc. Natl. Acad. Sci.,
98, p. 5481.
[33] Horinek, D. and J. Michl (2003) “Molecular dynamics simulation of an
electric field driven dipolar molecular rotor attached to a quartz glass sur-
face,” J. Am. Chem. Soc., 125, p. 11900.
[34] Zheng, H., M. E. Mulcahy, D. Horinek, F. Galeotti, T. F. Mag-
nera, and J. Michl (2004) “Dipolar and nonpolar altitudinal molecular
rotors mounted on an Au(111) surface,” J. Am. Chem. Soc., 126, p. 4540.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-99-320.jpg)
![85
[35] Clarke, L. I., D. Horinek, G. S. Kottas, N. Varaksa, T. F. Mag-
nera, T. P. Hinderer, R. D. Horansky, J. Michl, and J. P. Price
(2002) “The dielectric response of chloromethylsilyl and dychloromethylsilyl
dipolar rotors on fused silica surfaces,” Nanotehnology, 13, p. 533.
[36] Kottas, G. S., L. I. Clarke, D. Horinek, and J. Michl (2005) “Arti-
ficial molecular rotors,” Chem. Rev., 105, p. 1281.
[37] Jian, H. and J. M. Tour (2003) “En route to surface-bound electric field-
driven molecular motors,” J. Org. Chem., 68, p. 5091.
[38] Schmidt, M. W., K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S.
Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen,
S. Su, T. L. Windus, M. Dupuis, and J. A. Montgomery (1993)
“General atomic and molecular electronic structure system,” J. Comput.
Chem., 14, p. 1347.
[39] Gordon, M. S. and M. W. Schmidt (2005) “Advances in electronic struc-
ture theory: GAMESS a decade later,” in Theory and applications of com-
putational chemistry: the first forty years (C. E. Dykstra, G. Frenking, K. S.
Kim, and a G. E. Scuseria, eds.), Elsevier, Amsterdam, p. 1167.
[40] Li, H. and H. Jensen (2002) “Partial Hessian vibrational analy-
sis: the localization of the molecular vibrational energy and entropy,”
Theor. Chem. Acc., 107, p. 211.
[41] Head, J. D. (1997) “Computation of vibrational frequencies for adsorbates
on surfaces,” Int. J. Quant. Chem., 65, p. 827.
[42] Tu, Z. C. and X. Hu (2005) “Artificial molecular motor constructed
from a double-walled carbon nanotube driven by axially varying voltage,”
Phys. Rew. B, 72, p. 33404.
[43] Srivastava, D. (1997) “A phenomenological model of the rota-
tion dynamics of carbon nanotube gears with laser electric fields,”
Nanotechnology, 72, p. 186.
[44] Fennimore, A. M., T. D. Yuzvinsky, W. Han, M. S. Fuhrer,
J. Cummings, and A. Zetti (2003) “Rotational actuators based on carbon
nanotubes,” Nature, 424, p. 408.
[45] Zhang, S., W. K. Liu, and R. S. Ruoff (2004) “Atomistic simulations
of double-walled carbon nanotubes as rotational bearings,” Nanoletters, 4,
p. 293.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-100-320.jpg)
![86
[46] Zheng, Q. and Q. Jiang (2002) “Multiwalled carbon nanotubes as giga-
hertz oscillators,” Phys. Rev. Lett., 88, p. 45503.
[47] Dienwiebel, M., G. S. Verhoeven, N. Pradeep, and J. W. M.
Frenken (2004) “Superlubricity of graphite,” Phys. Rew. B, 92, p. 126101.
[48] Kolmogorov, A. N. and V. H. Crespi (2000) “Smoothest bearings:
Interlayer sliding in multiwalled carbon nanotubes,” Phys. Rew. Lett., 85, p.
4727.
[49] Legoas, S. B., V. R. Coluci, S. F. Braga, P. Z. Coura, S. O. Dan-
tas, and D. S. Galvao (2004) “Gigahertz nanomechanical oscillators based
on carbon nanotubes,” Nanotechnology, 15, p. 184.
[50] Bak, P. (1996) How nature works: The science of self-organised criticality,
Copernicus Press, New York, NY.
[51] Gruebele, M. (1998) “Intramolecular vibrational dephasing obeys a power
law at intermediate times,” Proc. Natl. Acad. Sci., 95, p. 5965.
[52] Drew, P. and L. Abbott (2006) “Models and properties of power-law
adaptation in neural systems,” J. Neurophysiol., 96, p. 826.
[53] Schreiber, F. (2000) “Structure and growth of self-assembling
monolayers,” Prog. Surf. Sci., 65, p. 151.
[54] Huang, L., G. Reekmans, D. Saerens, J. M. Friedt, F. Frederix,
L. Francis, S. Muyldermans, A. Campitelli, and C. Van Hoof
(2005) “Prostate-specific antigen immunosensing based on mixed self-
assembled monolayers, camel antibodies and colloidal gold enhanced sand-
wich assays,” Biosensors & Bioelectronics, 21, p. 483.
[55] Kwok, C. S., P. D. Mourad, L. A. Crum, and B. D. Ratner (2000)
“Surface modification of polymers with self-assembled molecular structures:
Multitechnique surface characterization,” Biomacromolecules, 1, p. 139.
[56] Mullen, T. J., A. A. Dameron, A. M. Andrews, and P. S. Weiss
(2007) “Selecting and driving monolayer structures through tailored inter-
molecular interactions,” Aldrichimica Acta, 40, p. 21.
[57] Mantooth, B. A. and P. S. Weiss (2003) “Fabrication, assembly, and
characterization of molecular electronic components,” Proc. IEEE, 91, p.
1785.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-101-320.jpg)
![87
[58] Moore, A. M., A. A. Dameron, B. A. Mantooth, R. K. Smith,
D. J. Fuchs, J. W. Ciszek, F. Maya, Y. X. Yao, J. M. Tour,
and P. S. Weiss (2006) “Molecular engineering and measurements to
test hypothesized mechanisms in single molecule conductance switching,”
J. Am. Chem. Soc., 128, p. 1959.
[59] Anderson, M. E., M. Mihok, H. Tanaka, L. P. Tan, M. W.
Horn, G. S. McCarty, and P. S. Weiss (2006) “Hybrid approaches
to nanolithography: Photolithographic structures with precise, controllable
nanometer-scale spacings created by molecular rulers,” Adv. Mater., 18, p.
1020.
[60] Dameron, A. A., J. R. Hampton, R. K. Smith, T. J. Mullen,
S. D. Gillmor, and P. S. Weiss (2005) “Microdisplacement printing,”
Nano Lett., 5, p. 1834.
[61] Dameron, A. A., J. R. Hampton, S. D. Gillmor, J. N. Hohman, and
P. S. Weiss (2005) “Enhanced molecular patterning via microdisplacement
printing,” J. Vac. Sci. Technol. B, 23, p. 2929.
[62] Smith, R. K., P. A. Lewis, and P. S. Weiss (2004) “Patterning self-
assembled monolayers,” Prog. Surf. Sci., 75, p. 1.
[63] Dameron, A. A., L. F. Charles, and P. S. Weiss (2005) “Struc-
tures and displacement of 1-adamantanethiol self-assembled monolayers on
Au(111),” J. Am. Chem. Soc., 127, p. 8697.
[64] Kassam, A., G. Bremner, B. Clark, G. Ulibarri, and R. B. Lennox
(2006) “Place exchange reactions of alkyl thiols on gold nanoparticles,”
J. Am. Chem. Soc., 128, p. 3476.
[65] Lussem, B., L. Muller-Meskamp, S. Karthauser, R. Waser,
M. Homberger, and U. Simon (2006) “STM study of mixed alka-
nethiol/biphenylthiol self-assembled monolayers on Au(111),” Langmuir, 22,
p. 3021.
[66] Schlenoff, J. B., M. Li, and H. Ly (1995) “Stability and self-exchange
in alkanethiol monolayers,” J. Am. Chem. Soc., 117, p. 12528.
[67] Dameron, A. A., T. J. Mullen, R. W. Hengstebeck, H. M.
Saavedra, and P. S. Weiss (2007) “Origins of displacement in
1-adamantanethiolate self-assembled monolayers,” J. Phys. Chem. C, 111,
p. 6747.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-102-320.jpg)
![88
[68] Mullen, T. J., A. A. Dameron, and P. S. Weiss (2006) “Directed
assembly and separation of self-assembled monolayers via electrochemical
processing,” J. Phys. Chem. B, p. 14410.
[69] Mullen, T. J., A. A. Dameron, H. M. Saavedra, M. E. Williams,
and P. S. Weiss (2007) “Dynamics of displacement in 1-adamantanethiolate
self-assembled monolayers,” J. Phys. Chem. C, 111, p. 6740.
[70] Poirier, G. E. (1997) “Characterization of organosulfur molecular mono-
layers on Au(111) using scanning tunneling microscopy,” Chem. Rev., 97, p.
1117.
[71] ——— (1997) “Mechanism of formation of Au vacancy islands in alkanethiol
monolayers on Au(111),” Langmuir, 13, p. 2019.
[72] Poirier, G. E. and E. D. Pylant (1996) “The self-assembly mechanism
of alkanethiols on Au(111),” Science, 272, p. 1145.
[73] Saavedra, H. M., C. M. Barbu, A. Dameron, T. J. Mullen, V. H.
Crespi, and P. S. Weiss (2007) “1-Adamantanethiolate monolayer dis-
placement kinetics follow a universal form,” J. Am. Chem. Soc., 129, p.
10741.
[74] Jensen, J. O. (2004) “Vibrational frequencies and structural determination
of adamantane,” Spectrochim. Acta, Part A, 60, p. 1895.
[75] Nuzzo, R. G., L. H. Dubois, and D. L. Allara (1990) “Fundamental-
studies of microscopic wetting on organic-surfaces.1. Formation and struc-
tural characterization of a self-consistent series of polyfunctional organic
monolayers,” J. Am. Chem. Soc., 112, p. 558.
[76] Bain, C. D. and G. M. Whitesides (1989) “Formation of monolayers by
the coadsorption of thiols on gold - variation in the length of the alkyl chain,”
J. Am. Chem. Soc., 111, p. 7164.
[77] Thomas, R. C., L. Sun, R. M. Crooks, and A. J. Ricco (1991) “Real-
time measurements of the gas-phase adsorption of normal-alkylthiol mono-
layers and multilayers on gold,” Langmuir, 7, p. 620.
[78] Karpovich, D. S. and G. J. Blanchard (1994) “Direct measurement
of the adsorption-kinetics of alkanethiolate self-assembled monolayers on a
microcrystalline gold surface,” Langmuir, 10, p. 3315.
[79] Langmuir, I. (1918) “The adsorption of gases on plane surfaces of glass,
mica and platinum,” J. Appl. Phys., 40, p. 1361.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-103-320.jpg)
![89
[80] Eberhardt, A., P. Fenter, and P. Eisenberger (1998) “Growth ki-
netics in self-assembling monolayers: a unique adsorption mechanism,” Surf.
Sci., 397, p. 285.
[81] Schwartz, P., F. Schreiber, P. Eisenberger, and G. Scoles (1999)
“Growth kinetics of decanethiol monolayers self-assembled on Au(111) by
molecular beam deposition: An atomic beam diffraction study,” Surf. Sci.,
423, p. 208.
[82] Peterlinz, K. A. and R. Georgiadis (1996) “In situ kinetics of self-
assembly by surface plasmon resonance spectroscopy,” Langmuir, 12, p.
4731.
[83] Dannenberger, O., M. Buck, and M. Grunze (1999) “Self-assembly
of n-alkanethiols: A kinetic study by second harmonic generation,” J. Phys.
Chem. B, 103, p. 22023.
[84] Woodward, J. T., I. Doudevski, H. D. Sikes, and D. K. Schwartz
(1997) “Kinetics of self-assembled monolayer growth explored via submono-
layer coverage of incomplete films,” J. Phys. Chem. B, 101, p. 7535.
[85] Spinke, J., M. Liley, F. J. Schmitt, H. J. Guder, L. Angermaier,
and W. Knoll (1993) “Molecular recognition at self-assembled monolayers
- Optimization of surface functionalization,” J. Chem. Phys., 99, p. 7012.
[86] Avrami, M. (1939) “Kinetics of phase change I - General theory,” J. Chem.
Phys., 7, p. 1103.
[87] ——— (1940) “Kinetics of phase change II,” J. Chem. Phys., 8, p. 212.
[88] ——— (1941) “Kinetics of phase change III,” J. Chem. Phys., 9, p. 117.
[89] Johnson, W. A. and R. F. Mehl (1939) “Reaction kinetics in processes
of nucleation and growth,” Trans. Am. Inst. Min. Metall. Eng., 135, p. 416.
[90] Holand, W., V. Rheinberger, and M. Schweiger (2003) “Control of
nucleation in glass ceramics,” Philos. Trans. R. Soc. Lond. Ser. A-Math.
Phys. Eng. Sci., 361, p. 575.
[91] Weinberg, M. C., D. P. Birnie, and V. A. Shneidman (1997) “Crys-
tallization kinetics and the JMAK equation,” J. Non-Cryst. Solids, 219,
p. 89.
[92] Shneidman, V. A. and M. C. Weinberg (1993) “The effects of tran-
sient nucleation and size-dependent growth-rate on phase-transformation
kinetics,” J. Non-Cryst. Solids, 160, p. 89.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-104-320.jpg)
![90
[93] Weinberg, M. and R. Kapral (1989) “Phase-transformation kinetics in
finite inhomogeneously nucleated systems,” J. Chem. Phys., 91, p. 7146.
[94] Jun, S., H. Y. Zhang, and J. Bechhoefer (2005) “Nucleation and
growth in one dimension. I. The generalized Kolmogorov-Johnson-Mehl-
Avrami model,” Phys. Rev. E, 71, p. 11908.
[95] Kooi, B. J. (2004) “Monte Carlo simulations of phase transformations
caused by nucleation and subsequent anisotropic growth: Extension of the
Johnson-Mehl-Avrami-Kolmogorov theory,” Phys. Rev. B, 70, p. 224108.
[96] ——— (2006) “Extension of the Johnson-Mehl-Avrami-Kolmogorov theory
incorporating anisotropic growth studied by Monte Carlo simulations,” Phys.
Rev. B, 73, p. 54103.
[97] Holloway, P. H. and J. B. Hudson (1974) “Kinetics of reaction of oxygen
with clean nickel single-crystal surfaces. 1. Ni(100) surface,” Surf. Sci., 43,
p. 123.
[98] ——— (1974) “Kinetics of reaction of oxygen with clean nickel single-crystal
surfaces. 2. Ni(111) surface,” Surf. Sci., 43, p. 141.
[99] Okada, T., W. Utsumi, H. Kaneko, V. Turkevich, N. Hamaya, and
O. Shimomura (2004) “Kinetics of the graphite-diamond transformation in
aqueous fluid determined by in-situ X-ray diffractions at high pressures and
temperatures,” Phys. Chem. Miner., 31, p. 261.
[100] Ruitenberg, G., A. K. Petford-Long, and R. C. Doole (2002) “De-
termination of the isothermal nucleation and growth parameters for the crys-
tallization of thin Ge2Sb2Te5 films,” J. Appl. Phys., 92, p. 3116.
[101] Baird, J. K., S. C. Hill, and J. C. Clunie (1999) “Kinetics of protein
crystal nucleation and growth in the batch method,” J. Cryst. Growth, 196,
p. 220.
[102] Fanfoni, M. and M. Tomellini (1998) “The Johnson-Mehl-Avrami-
Kolmogorov model: a brief review,” Il Nuovo Cimento, 20, p. 1171.
[103] Ercolani, G. (2005) “The origin of cooperativity in double-wheel receptors.
Freezing of internal rotation or ligand-induced torsional strain?” Org. Lett.,
7, p. 803.
[104] Sugasaki, A., M. Ikeda, M. Takeuchi, A. Robertson, and S. Shinkai
(1999) “Efficient chirality transcription utilizing a cerium(IV) double decker
porphyrin: a prototype for development of a molecular memory system,” J.
Chem. Soc., 1, p. 3259.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-105-320.jpg)
![91
[105] Tashiro, K., K. Konishi, and T. Aida (2000) “Metal bisporphyrinate
double-decker complexes as redox-responsive rotating modules. Studies on
ligand rotation activities of the reduced and oxidized forms using chirality
as a probe,” J. Am. Chem. Soc., 122, p. 7921.
[106] Bian, Y., D. Wang, R. Wang, L. Weng, J. Dou, D. Zhao, D. Ng,
and J. Jiang (2003) “Structural studies of the whole series of lanthanide
double-decker compounds with mixed 2,3-naphthalocyaninato and octaethyl-
porphyrinato ligands,” New J. Chem., 27, p. 844.
[107] Bian, Y., R. Wang, D. Wang, R. Wang, P. Zhu, R. Li, J. Dou,
W. Liu, C. Choi, H. Chan, C. Ma, D. Ng, and J. Jiang (2004)
“Synthesis, structure, and spectroscopic and electrochemical properties
of heteroleptic bis(phthalocyaninato) rare earth complexes with a C4
symmetry,” Helv. Chim. Acta, 87, p. 2581.
[108] Tashiro, K., K. Konishi, and T. Aida (1997) “Enantiomeric resolu-
tion of chiral metallobis(porphyrin)s: Studies on rotatability of electronically
coupled porphyrin ligands,” Angew. Chem. Int. Ed. Engl., 36, p. 856.
[109] Takami, T., D. Arnold, A. Fuchs, G. Will, R. Goh, E. Waclawik,
J. Bell, P. Weiss, K. Sugiura, W. Liu, and J. Jiang (2006) “Two-
dimensional crystal growth and stacking of bis(phthalocyaninato) rare earth
sandwich complexes at the 1-phenyloctane/graphite interface,” J. Phys.
Chem. B, 110, p. 1661.
[110] Takami, T., T. Ye, D. Arnold, K. Sugiura, R. Wang, J. Jiang,
and P. Weiss (2007) “Controlled adsorption orientation for double-decker
complexes,” J. Phys. Chem. C, 111, p. 2077.
[111] Ye, T., T. Takami, R. Wang, J. Jiang, and P. Weiss (2006) “Tuning
interactions between ligands in self-assembled double-decker phthalocyanine
arrays,” J. Am. Chem. Soc., 128, p. 10984.
[112] Strogatz, S. (2001) Nonlinear dynamics and chaos, John Wiley & Sons,
Boulder, Colorado.](https://image.slidesharecdn.com/2b7ad874-486d-411f-bb92-7946b715d78e-151127113605-lva1-app6892/85/Corina_Barbu-Dissertation-106-320.jpg)













































