
![Hunter 2
Abstract
The gpD capsid protein of bacteriophage λ has been used for efficient display of fusion
proteins at both the C and N terminus, generating Lambda Display Particles (LDP). Previous
work to complement i434Dam123 phage with a library of gpD-fusion expressing strains produced
limited results. An attempt was made to isolate mutants of i434Dam123 that could be
complemented more readily by the gpD-fusions. It was hypothesized that by selecting for
missense mutants of Dam123, revertants could be selected that have greater tolerance for plating
on different D-fusions. i434Dam123 was plated on 594 and 594[p617] to generate mutants that
could not plate when gpD-fusions were exogenously expressed from 594[p617]. The resulting
mutants could not plate on 594[p617], and were termed “IPDF”mutants. These were then used
to isolate suppressor mutants that regained the ability to plate equally on both 594 and
594[p617], termed “supIPDF” mutants. Sequencing of D and E for both classes of mutants
determined that they were true revertants to D+, and the mutation(s) responsible for the IPDF and
supIPDF phenotypes were extragenic to D and E. Furthermore, supIPDF mutants were shown to
equally plate in the presence or absence of the p626 gpD-fusion, suggesting that gpD provided
by the phage was sufficient for plating, and that gpD-fusion decoration was not obligatory for
plating. The results suggest that there are mutations extragenic to D and E that can both enhance
and suppress toxicity of a given gpD-fusion towards viable phage assembly.
Introduction
Phage display is a relatively recent technology that involves the expression of foreign
proteins on the surface of a bacteriophage. Phage display was first discovered in 1985 by
George Smith, who successfully displayed EcoR1 on pIII in filamentous phage (Smith, 1985).](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-2-320.jpg)


![Hunter 5
sequencing, the Qiagen PCR Purification kit was used with provided buffers (PB buffer, EB
elution buffer, loading dye, 1kb DNA ladder). For the marker rescue assay, TM buffer (0.01M
Tris, 0.01M MgSO4, dH20, adjusted to pH=7.6) was used for starving of the cells.
In vivo complementation assay
For plating of i434Dam123 and i434(1812)p22 phage on fusion host strains to assess
complementation, phage culture dilutions were prepared to 10-7 in Φ80 buffer. Fresh overnight
cells of the 594[D-fusion] and TC600[D-fusion] strains were grown overnight in LB+Amp50.
At incubation temperatures of 25ºC and 30ºC, plating dilutions of 10-2, 10-3, and 10-4 were used.
0.3ml of the cells were added to each of the plating dilution tubes. 0.1ml of the 10-1, 10-2, and
10-3 culture dilutions were then added to the plating dilution tubes. At incubation temperatures
of 37ºC and 41ºC, 10-5, 10-6, and 10-7 plating dilution tubes were used. 10-2, 10-3, and 10-4
plating dilution tubes were used for plating the tester phage on non-permissive 594 cells. 10-6
and 10-7 tester phage were plated on permissive supE TC600 cells. Plating on 594 and TC600
was grown overnight at 30ºC to calculate reversion frequency.
Primary phage lysate preparation
Primary phage lysates were isolated from a single plaque stripped out on a permissive
host strain. 20ml of LB was added to 0.2ml of 1M MgCL2, 0.2ml of 1M CaCl2, and 0.2ml of 1M
Tris in a 125ml flask. The contents were then placed in a 37ºC shaking waterbath. For coring of
the plaques, 0.5ml of Φ80 buffer was added to a 1.5ml microfuge tube. 1 plaque from the strip
plate was cored out using an Aardvark pipette and added to the microfuge tube. The contents
were finger vortexed and incubated in the 39ºC incubator for 15min to allow for phage diffusion.
After the 15min incubation period, 0.2ml of 0.02M CaCl2+MgCl2 solution and 0.2ml of indicator](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-5-320.jpg)

![Hunter 7
2min. 0.8ml of LB media was then added to the mixture to a total volume of 1ml. The mixture
was then incubated in the 37ºC shaking waterbath for 60min.
Stripping phage lysates for single plaques
For stripping of phage lysates onto an indicator strain to isolate single plaques, 0.3ml of
the indicator cells with 3ml of LB top agar was poured onto a plate and cooled. 50µl of phage
lysate was then pipetted onto the indicator plate and streaked with sterile paper strips.
Isolation of IPDF mutants
To isolate phage mutants capable of enhanced decoration with exogenous gpD-fusions, a
mutant strain with inhibited plating on gpD-fusions (IPDF) was first isolated. This would
provide a selective pressure for isolating the supIPDF mutants. To isolate the IPDF mutants,
i434Dam123 phage were plated at a plating dilution of 10-2 on 594 at 38ºC. The high titre plating
on the permissive 594 strain would allow Dam123 revertants capable of plating on 594 to be
selected. At a plating dilution of 10-2, 72 plaques were present on 594. 20 of these plaques were
then picked onto 594 and 594[p617]. Of the 20 plaques that were picked, A-1, A-5, B-4, D-1,
and D-5 showed +1 (2-3mm) or (-) (no plaque) plating on 594[p617] and +3 (>3mm) plating on
594, indicating that the gpD-fusion was inhibiting plating. The plaques of each of these mutants
were cored from the 594 plate into 0.5ml of Φ80 buffer and stripped onto 594 and 594[p617].
For each one of the mutants, 10 plaques were picked directly from the strip plate onto 594 and
594[p617] separately. The 10 plaques picked were divided into two sets of 5 plaques stabbed
each onto 594 and 594[p617]. The reference system for stabbing the plaques consisted of set 1
having 5 plaques from each strip plate stabbed onto 594 and 594[p617] and labelled O, N, M, L,
K. For set 2, 5 plaques from each strip plate was stabbed onto 594 and 594[p617] and designated](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-7-320.jpg)
![Hunter 8
T, S, R, Q, and P. The results of the plaque picking assay showed extensive lysis on 594 with no
lysis on 594[p617] except for D-1, A-5-m, and A-5-p. To make primary phage lysates from the
mutants, one plaque for each of the mutants was selected from the “t” column of the 594 plate
from set 2. This plaque was cored in Φ80 buffer and stripped onto 594 and 594[p617] to purify
the phage. The strip plates agreed with the previous plaque picking assay, showing extensive
lysis on 594 plates with no lysis on 594[p617] apart from D-1. From each of the 594 strip plates,
plaques from each mutant were cored and purified for plaque PCR and sequencing of the D and
E regions. Primary phage lysates were made for each mutant, with 3 plaques cored from each
mutant in Φ80 buffer for making a primary lysate using 594. The primary lysates for each
mutant were then titred on 594 and 594[p617] to verify the IPDF phenotype. The resulting titres
indicated a 107 fold increase in plating on 594 compared to 594[p617], indicating a strong IPDF
phenotype. The resulting IPDF lysates were labelled IPDF A1, IPDF A5, IPDF B4, IPDF D5,
and IPDF D1 as a revertant control expressing no actual IPDF phenotype.
Isolation of supIPDF mutants
To isolate the supIPDF mutants from the IPDF lysates, 10-1 plating dilutions of each
IPDF lysate was plated onto 594 and 594[p617] and incubated at 38ºC. The resulting plaques on
594[p617] were original IPDF mutants that had gained a secondary mutation that negated their
IPDF phenotype. 10 plaques from IPDF A1, A5, D5, 15 plaques from IPDF B4, and 5 plaques
from IPDF D1 were picked from their respective 594[p617] plate and stabbed onto 594[p617].
All of the plaques from this picking assay that exhibited +3 plating on 594[p617] were then
further stabbed onto 594 and 594[p617]. The total number of plaques stabbed onto 594 and
594[p617] from all IPDF lysates was 28. Of the 28 plaques stabbed onto 594 and 594[p617], 4
were selected for preparing primary phage lysates due to consistent plaque morphology. These 4](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-8-320.jpg)
![Hunter 9
plaques were cored from B4-2-2, D1-1-1, D5-1-1, and D5-2-2 becoming supIPDF B4, D1, D5-1,
and D5-2 respectively. The primary phage lysates were made by coring the 4 mutant plaques
from the 594[p617] plate in Φ80 buffer and stripping onto 594. From the 594 strip plates, the
primary phage lysates were then made and titred on 594 and 594[p617]. All of the supIPDF
lysates exhibited 109 plating on both 594 and 594[p617].
Plaque PCR of IPDF mutants (performed by Dr. Hayes)
To sequence D in IPDF mutants, 2 plaques from each of the 594 strip plates were cored
out and added to 0.1ml of TE buffer. Samples were incubated in a 37ºC heating block for 20min,
followed by incubation in the 37ºC incubator for 90min. After incubation, each of the respective
IPDF samples were transferred to a 96ºC heating block for 5min and then added to the PCR
reaction mixture:
10µl plaque solution heated 5min at 96ºC
10µl 10X ThermoPol buffer with MgCl2 *11= 110µl
16µl 1.25mM dNTPs *11=176µl
2.5µl Primer #1 L-Nu3forD at 40µM *11= 27.5µl
2.5µl Primer #4 RendEforD at 40µM *11= 27.5µl
58.5µl dH2O *11= 643.5µl
0.5µl Taq DNA Polymerase at 5µ/ml
Each reaction tube had a total volume of 100µl, with 10 reaction tubes in total (2 tubes
for each of the IPDF plaques). 28 cycles were run at an annealing temperature of 48ºC. After
PCR, 90µl of each sample was frozen, and the remaining reaction mixture was run on a 0.8%
agarose gel with 4µl of loading dye added to each sample. 24-25µl of each reaction sample was](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-9-320.jpg)

![Hunter 11
58.5µl dH2O *7= 409.5µl
0.5µl Taq DNA Polymerase at 5µ/ml
Samples that were being amplified for D were mixed with Primer #1 LNu3forD and
Primer #4 RendEforD, and samples amplified for E were mixed with Primer #2 LDforE and #5
RfiforE. Plaques were picked from 594 strip plates grown at 30ºC. The PCR reaction was
carried out at an annealing temperature of 48ºC. PCR products were run on a 0.8% Agarose gel
with 10µl of sample, 10µl of TE buffer, and 2µl of loading dye for each supIPDF amplified
product. As a reference, 18µl TE buffer was added to 2µl of 1kb DNA ladder and 2µl of loading
dye. Each of the supIPDF samples for amplification of D and E were run in duplicate on a gel.
Each of the duplicate samples were then pooled and purified using a Qiagen PCR Purification
kit. For both IPDF and supIPDF lysates, sequencing data was analyzed using SnapGene
software version 2.7.2 (snapgene.com).
Testing for virλ contaminants in #1027 i434Dam123
To ensure that IPDF and supIPDF mutants were not initially derived from low-level virλ
contaminants present in the i434Dam123 lysate, plating on different indicator host strains was
carried out to verify the immunity regions. For comparison, MMS179 #1027b i434Dam123 was
used as a reference control. This strain was verified by Connie Hayes in previous experiments.
Each of the i434Dam123 strains was plated on TC600, 594, TC600[λpapa] and TC600[i434T].
Plating dilutions for TC600[i434T] and 594 were done at 10-2, 10-3, and 10-4 because both strains
are non-permissive to i434Dam123, and low plating dilutions would allow detection of
contaminating λvir. Plating dilutions for TC600 and TC600[λpapa] were carried out at 10-6, 10-7,](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-11-320.jpg)
![Hunter 12
and 10-8. Plates were incubated overnight at 30ºC, and the titres of 594 and TC600 were used to
calculate the reversion frequency.
Testing for cI857 repressor effects for supIPDF plating on gpD-fusion strains at 30ºC and
40ºC
To determine what effects the cI857 repressor had on the ability of supIPDF lysates to
plate on gpD-fusion strains, plating was carried out on 594[p613], 594[p613*], and 594[p626] at
30ºC and 40ºC. Previous data indicates that cI857 repressor is bound at operator sites at 30ºC.
This would prevent exogenous expression of gpD-fusions in the host. At 40ºC, cI857 is
completely derepressed, and there is full gpD-fusion expression. 594[p613] was chosen as a
positive control, with WT gpD expression at 40ºC. 594[p613*] was chosen as a negative
control, having no gpD expression at both temperatures. Plating dilutions of 10-7 and 10-8 were
used for all supIPDF lysates. The fold change in plating was calculated using the titres of the
supIPDF mutant at 30ºC and 40ºC.
Marker rescue assay
To determine if low level plating of supIPDF lysates on fusion strains at 30ºC was due to
recombination between Dam123 and D+, a marker rescue assay was carried out. By producing
lysates of “input” and “output” phage and then comparing the level of plating on a permissive
and non-permissive host, the level of plating due to marker rescue was determined. 2mls of
overnight cultures for 594, TC600, 594[p613], and 594[p613*] were inoculated into 3ml of TM
buffer, with 10-7 and 10-8 of cells being spread on TC600 to determine CFU before the cells were
diluted. The inoculated cultures in TM buffer were then kept at room temperature for 1 hour.
0.1ml of the starved cells were then transferred to 30.86ml of TM buffer to obtain a culture of](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-12-320.jpg)
![Hunter 13
1.62*107 CFU/ml. The starved cells were then infected by #1027 i434Dam123 phage at an MOI
of 5 in a 1.5ml microfuge tube by adding 0.1ml of phage to 1ml of cells. The infected cultures
were then incubated in a 39ºC heating block for 15min. Infected cultures were then transferred
to 20ml of prewarmed LB at 39ºC and incubated in a 39ºC shaking water bath for 90min.
Several drops of Chloroform were then added to the cultures and further shaken for 5min.
Lysates were centrifuged at 8k for 10min and then transferred to lysate bottles. These lysates
were then plated 10-1, 10-2, 10-3 on 594 and 10-4, 10-5, 10-6 on TC600 to calculate the marker
rescue frequency of the “output” phage. For comparison, #1027 i434Dam123 phage was plated
on 10-1, 10-2, and 10-3 on 594 and 10-6, 10-7, and 10-8 on TC600 to calculate the reversion
frequency of the “input” phage.
Testing for iλDam123 contaminants in #1027 i434Dam123 lysate
To determine if there were any iλDam123 contaminants in the lysate used for isolating the
IPDF and supIPDF lysates, #1027 i434Dam123 was plated 10-2 and 10-3 on 594 and
TC600[i434T]. The lysate was also plated on TC600 and TC600[λpapa] at plating dilutions of
10-7 and 10-8. The plates were incubated overnight at 30ºC.
Results
In vivo complementation data compiled over the summer indicated that using the
i434Dam123 strain for exogenous gpD-fusion complementation was problematic for most 594
gpD-fusion strains. The exceptions to this were 594[p613] and 594[p614] expressing WT gpD
and an N-terminal His-gpD fusion respectively (Table 5). The TC600 gpD-fusion strains all
showed plating at high titres for both permissive and non-permissive temperatures, indicating
that the nonsense suppressor tRNA of the SupE strain compensated the amber mutation in D.](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-13-320.jpg)
![Hunter 14
Given that the only strains able to complement i434Dam123 were the positive control and a His-
gpD fusion, the results indicated that i434Dam123 was incapable of being complemented by gpD-
fusions larger than 11aa. Possible reasons for this could be the fact that there was no ability of
the expressed gpD-fusion protein to interact with the truncated gpD in forming an effective
trimer on the capsid, or that there were gpD-fusion-truncated gpD trimers produced that
destabilized the capsid. Also, it was shown that in all 594 gpD-fusion hosts, there was low-level
plating of i434Dam123 at 30ºC. Because the cI857 repressor bound to the operator sites of the
plasmid at 30ºC are not derepressed, it was deemed not possible that the plating could be due to
gpD-fusion expression. It was hypothesized that the low-level plating could be due to marker
rescue between D+ and Dam123.
To test whether marker rescue was responsible for the low-level plating at 30ºC, output
phage grown on 594[p613] were plated on 594 and TC600, and the frequency of plating was
compared to the reversion frequency of the input phage, i434Dam123. The results showed that
there was a 273% increase in the efficiency of plating (EOP) of the output phage compared to the
input phage, suggesting that marker rescue took place (Table 25).
To isolate λ mutants with improved plating on 594 gpD-fusion hosts, mutants with
inhibited plating in the presence of gpD-fusions first had to be isolated. This provided a counter
selection for selecting mutants with increased gpD-fusion display. To isolate mutants with
inhibited plating on gpD-fusions, i434Dam123 was first plated on 594 to isolate mutants with
reversions and mismatch mutations in Dam123. The resulting plaques were then picked onto
594[p617], a host with a plasmid encoding for a 155aa N-terminal gpD-fusion (Table 1). By
picking onto 594[p617], it was hypothesized that missense mutants with decreased plating on the
gpD-fusion host could be selected. The results showed that of the 20 plaques isolated on 594, 5](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-14-320.jpg)
![Hunter 15
showed decreased plaque sizes on 594[p617] (Table 7). These 5 plaques were then further
isolated by picking onto plates in two sets, with each set involving plaques being picked onto
594 and 594[p617]. This ensured that the plaques picked were not being selected from
594[p617], and were only plaques from the phage capable of plating on 594 (Table 8). From the
5 mutants in the “T” column of set 2, primary lysates were made and titred on 594 and
594[p617]. The resulting IPDF lysates (inhibition of plating on D-Fusions) were shown to have
high titre plating on 594 with little to no plating on 594[p617] (Table 9). The exception to this
was IPDF D1, which showed high titre plating on both 594 and 594[p617]. This mutant was
therefore not actually an “IPDF” mutant, but was considered for further selection as a positive
control. IPDF D1 also produced turbid plaques on 594 and 594[p617], whereas the other IPDF
mutants produced clear plaques.
To isolate λ mutants with improved plating on 594 D-fusions using the IPDF lysates as a
counter selection, each of the IPDF lysates were plated 10-1 on 594[p617] to generate supIPDF
mutants. 10-15 plaques from each 594[p617] plate were then stabbed onto 594[p617] to further
purify the phage, with supIPDF plaques being generated from IPDF D1, IPDF B4, and IPDF D5.
The results show that the IPDF lysates produced supIPDF lysates at different frequencies, with
IPDF A1 and IPDF A5 producing no supIPDF mutants, and supIPDF D5 producing 10 (Table
10). This suggests that suppressor mutations of IPDF mutants arise at different rates, indicating
that the mutations that give rise to the IPDF mutants are not all the same. Furthermore, stabbing
the putative supIPDF mutants from 594[p617] again onto 594 and 594[p617] produced clear and
turbid plaques in some of the plaques picked onto 594, while all the stabs on 594[p617] were
turbid (Table 11 and 12). It is possible that this difference is due to the mixture of supIPDF and
IPDF mutants within a single plaque that is picked. IPDF mutants that do not grow on the](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-15-320.jpg)
![Hunter 16
594[p617] host but remain on the plate could be picked up and transferred to 594 to produce
clear plaques. This is further supported by the fact that supIPDF mutants arising from IPDF D1
do not produce clear plaques. Given that all progeny phage arise in a plaque from a single phage,
it is unlikely that the difference in plaque morphology is due to genetic instability in the progeny
phage. Furthermore, the presence or absence of expressed gpD-fusion in 594[p617] should not
have any impact on plaque morphology. Of the 23 plaques picked onto 594 and 594[p617], 4
were selected for purification as supIPDF mutants. Primary lysates of these mutants were made,
and resulted in high and equal titres on 594 and 594[p617] at 38ºC (Table 13).
To assess whether cI857 repressor activity had any effect on the plating of supIPDF
lysates through abrogation of exogenous gpD/gpD-fusion expression, the supIPDF lysates were
plated on 594[p613] and 594[p613*] at 30ºC and 40ºC. Since supIPDF lysates were capable of
plating on 594 in the absence of gpD/gpD-fusion expression, it was important to determine if
gpD/gpD-fusion expression improved or abrogated supIPDF plating. The results showed that in
the presence of gpD from 594[p613], plating increased 1.14-1.89 fold (Table 20). By
comparison, plating was increased 1.28-3.17 fold with no gpD expression from 594[p613*] at
40ºC. This indicated that supIPDF mutants plated independently of exogenous gpD/gpD-fusion
expression (Table 21).
To test whether IPDF or supIPDF mutants originated from λvir contaminants in the
i434Dam123 culture, #1027 i434Dam123 was plated at low plating dilutions on known permissive
and non-permissive hosts carrying different immunity regions to detect the presence of virλ. The
assay was run in parallel with MMS 179 #1027b i434Dam123, an identical strain previously
verified to have no λvir contaminants. The results showed the presence of two possible plaques](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-16-320.jpg)
![Hunter 17
at 10-2 plating dilutions on TC600[i434T] (Table 15 and 16). These plaques were not at a high
enough concentration to account for the IPDF mutants isolated from i434Dam123.
Isolation of supIPDF mutants was initially thought to be due to selecting for missense
mutations of Dam123 that resulted in more stable gpD-gpD-fusion interactions. To determine
the genotype of the IPDF and supIPDF mutants, D and E for each class of mutants was amplified
in plaque PCR and sequenced. The results show that both IPDF and supIPDF mutants reverted
to WT D, and E for both classes of mutants were WT (Tables 17-19). Sequencing results for the
D of the IPDF mutants were analyzed by Dr. Hayes, and have not been included in a table for
this report. The sequencing data indicated that there were no missense mutations in D that
improved or inhibited the level of plating on 594 gpD-fusions, but that the mutation(s) that
caused the phenotypes were extragenic to D and E.
Discussion
The plating data for the IPDF mutants indicates an inhibitory mutation that prevents
plating on 594[p617] despite wildtype expression of both gpD and gpE. This inhibitory mutation
became suppressed in the supIPDF mutants, allowing high titre and equal plating on both
594[p613*], 594[p613], and 594[p626] (Table 20-22). Based on these observations, it is
important to determine at what frequency the supIPDF mutants were isolated from the IPDF
mutants. Because there were uncountable pinpoint plaques when IPDF lysates were plated on
594[p617], only conservative estimates can be calculated for the frequency of supIPDF mutant
isolation. Based on the titres of the IPDF mutants on 594 (Table 14) and the fact that they were
each plated on 594[p617] at a plating dilution of 10-1, approximately 5.4*108, 7.9*108, and
2.4*108 PFU were present on the 594[p617] plate for IPDF D5, IPDF B4, and IPDF D1](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-17-320.jpg)
![Hunter 18
respectively. Assuming that each one of the plaques with +3 plating on 594[p617] was a
putative supIPDF mutant, than each supIPDF mutant was selected from a pool of putative
supIPDF mutants derived from a single IPDF mutant. As a result, the frequency for selecting
supIPDF B4 from IPDF B4 would be 8/7.9*108= 1*10-8. For selecting supIPDF D5-1 and
supIPDF D5-2 from IPDF D5, the frequency would be 10/5.4*108= 1.85*10-8. Given that IPDF
D1 had high titre plating on both 594 and 594[p617], isolation of a supIPDF mutant was not
possible because there wasn’t an actual “IPDF” phenotype to begin with. Despite this, IPDF D1
and supIPDF D1 remained as misnomers for the purpose of determining if there were any
mutations different from the actual IPDF and supIPDF mutants.
It is also important to note that in the case of IPDF A1 and IPDF A5, no supIPDF
mutants were isolated. This indicated that there were different and perhaps multiple mutations
that cause the IPDF phenotype. If generation of a supIPDF phenotype is due to reversion of the
IPDF mutation, having more than one mutation causing the IPDF phenotype would cause the
strain to revert to the supIPDF phenotype at a significantly lower frequency. Likewise, if the
supIPDF phenotype is due to a suppressor mutation, having more than one mutation responsible
for the IPDF phenotype would decrease the likelihood of generating suppressor mutations
against all of the different IPDF mutations. The potential variability in number and types of
mutations that could be responsible for the IPDF phenotype would not be surprising considering
the high degree of specificity in protein-protein interactions during λ capsid assembly.
Based on the genetic data showing wildtype D and E for both mutant strains (Table 17-
19), and the plating data showing high level plating for both mutant strains on 594 (Table 14), it
is likely that supIPDF and IPDF mutants do not require gpD-fusion involvement during capsid
assembly given the high titre plating in the absence of gpD-fusion expression. The phenotypic](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-18-320.jpg)

![Hunter 20
while gpD-fusion plating data for the IPDF mutants is restricted only to 594[p617] and
TC600[p617], the supIPDF mutants that were isolated exhibited a supIPDF phenotype against a
different fusion strain, 594[p626]. This indicates that while there is plating data lacking for
IPDF and supIPDF plating on other gpD-fusion strains, the plating data of supIPDF mutants
suggests that the supIPDF phenotype is applicable to other gpD-fusion strains besides the strain
that the mutants were isolated from. A possible explanation for this could be the fact that p617
and p626 gpD-fusions both express gpD-fusions on the N-terminus. It remains to be seen if
plating on gpD-fusions expressed from the C-terminus would still confer the supIPDF
phenotype.
The recalcitrant nature of gpD-fusions can also explain the reason why in vivo
complementation assays using i434Dam123 were largely unsuccessful (Tables 5 and 6). The
difference in the level of plating between the permissive TC600 and non-permissive 594 gpD-
fusion expressing strains indicates that any plating at all was only possible with wildtype gpD
present. This is supported in the literature, with λ decoration solely from gpD-fusions causing
interference in capsid formation and assembly (Zanghi et al, 2005). The exception to this was
the presence of high titre plating at 37ºC and 41ºC on 594[p614] (Table 5). The p614 plasmid
encodes for an 11aa N-terminal His tag (Table 1). The small size of this gpD-fusion is in
agreement with previous experiments suggesting that the smaller the size of the fusion protein,
the greater the number of gpD-fusions that can be expressed on the phage as a result of increased
stability (Gupta et al, 2003). Certainly, this would be the size limit for expression on
i434Dam123, given that all other gpD-fusions were incompatible. It seems that having a correct
ratio of wildtype gpD to gpD-fusion is necessary for the stability of the gpD trimer, and effective
stabilization of the expanded procapsid. This has been previously determined experimentally,](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-20-320.jpg)
![Hunter 21
with the use of different suppressor tRNA expressing host strains to regulate the ratios of gpD
and gpD-fusion expression for optimal phage display (Mikawa et al, 1996). The instability
generated by p617 D-fusions can also be explained given that the 46aa addition is on the N-
terminal end. Despite the fact that an N-terminal linker was added to the construct in an attempt
to prevent interference of gpD trimer formation by the N-terminal fusion, it is likely that the N-
terminal addition still caused unfavourable interactions with gpE. This is also supported by
cryo-electron microscopy showing that the first 14 residues in the gpD N-terminus interact with
an E-loop strand in gpE to form a β sheet (Lander et al, 2008). Having the gpD trimers
consisting of 100% gpD-fusions can cause these unfavourable interactions, necessitating control
mechanisms to regulate the expression of wildtype gpD and gpD-fusion, which were not
sufficient for the system used in this study.
Given the mostly negative results for in vivo complementation on the 594 gpD-fusion
host strains (Tables 5 and 6), the presence of plating at 30ºC on the gpD fusion strains by
i434Dam123 would have to be due to wildtype gpD, either as a result of reversion or marker
rescue. To test whether marker rescue or reversion was responsible, a marker rescue assay was
performed. The results of the assay showed that the frequency of marker rescue on 594[p613]
was over 2 fold higher than the reversion frequency for i434Dam123 (Table 25). Plating the
secondary lysate grown on 594[p613*] onto 594 produced no revertants, indicating that the low
titre plating was not due to reversion (Table 23). This minor increase above the reversion
frequency of i434Dam123 also explains why plating at 30ºC and sometimes 25ºC produced only
103-4 phage despite the titre of i434Dam123 on TC600 being 109.
This study has shown that missense mutants of i434Dam123 capable of efficacious
decoration by various gpD-fusion proteins are not selectable using a selection scheme on a non-](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-21-320.jpg)
![Hunter 22
permissive 594 and 594[p617] host. Instead, it is much more likely that selection of wildtype
gpD revertants will occur along with mutations extragenic to D and E that cause interference by
p617 gpD-fusion, and removal of interference by the same and other similar gpD-fusions. The
presence of wildtype D and E in both the IPDF and supIPDF mutants further highlight that gpD-
gpE interactions are very important for the stability of the capsid, and extragenic mutations that
impede this interaction through preferential binding of gpD-fusions are toxic to the phage. This
problem is an example of why regulation of gpD and gpD-fusion expression is so important. In
the system used for this project, gpD-fusion expression from the pcIpR plasmid was under the
control of cI857, a temperature sensitive cI repressor that has been shown in previous
experiments to effectively control expression of protein P in 594[pcIpR-P-timm] (Hayes et al,
2013). Despite this, there was no regulation in place for wildtype gpD expression, which has
been proven essential in displaying fusion proteins through facilitating cooperative binding
(Yang et al, 2000). Instead, it was expected that different missense mutations of Dam123 would
be selected, and some would be more conducive to gpD-fusion complementation than others. In
another experiment, missense mutations in λDam15 mutants were generated through the use of
plating on different isogenic suppressor strains (Nicastro et al, 2013). The experiment allowed
control not only of the level of gpD-fusion expression, but also the types of missense mutants
that were generated in Dam15. This approach addressed a shortcoming made evident in this
study, in that missense mutations in Dam15 were directly selected through the use of the isogenic
suppressors, reducing reversion to the wildtype and production of other mutations. The results of
this study indicate that these mutations were extragenic to both D and E, and conferred
enhancement and suppression of toxicity by a gpD-fusion towards viable phage assembly.](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-22-320.jpg)

![Hunter 24
Table 2: Bacterial strains
Name Genotype Source
B10 594 F- lac-3350 galK2 galT22 rpsL179 IN(rrnD-rrnE)1; Hayes Lab
B8 TC600 supE, Pm+ Hayes Lab
TC600[i434T] supE, Pm+, λimm434T lysogen Hayes Lab
TC600[λpapa] supE, Pm+, λpapa lysogen Hayes Lab
594[p613] as before, transformed with p613 plasmid Hayes Lab
TC600[p613] as before, transformed with p613 plasmid Hayes Lab
594[p613*] as before, transformed with p613* plasmid Hayes Lab
TC600[p613*] as before, transformed with p613* plasmid Hayes Lab
594[p614] as before, transformed with p614 plasmid Hayes Lab
TC600[p614] as before, transformed with p614 plasmid Hayes Lab
594[p614*] as before, transformed with p614* plasmid Hayes Lab
TC600[p614*] as before, transformed with p614* plasmid Hayes Lab
594[p617] as before, transformed with p617 plasmid Hayes Lab
TC600[p617] as before, transformed with p617 plasmid Hayes Lab
594[p623] as before, transformed with p623 plasmid Hayes Lab
TC600[p623] as before, transformed with p623 plasmid Hayes Lab
594[p625] as before, transformed with p625 plasmid Hayes Lab
TC600[p625] as before, transformed with p625 plasmid Hayes Lab
594[P626] as before, transformed with p626 plasmid Hayes Lab
TC600[p626] As before, transformed with p626 plasmid Hayes Lab
Table 3: Phage strains
Name Source
#1027 imm434Dam123 Hayes Lab
MMS 179 #1027b imm434Dam123 Hayes Lab
imm434(18,12)p22 Hayes Lab
IPDF A1 Hayes Lab
IPDF A5 Hayes Lab
IPDF B4 Hayes Lab
IPDF D1 Hayes Lab
IPDF D5 Hayes Lab
supIPDF B4 Hayes Lab
supIPDF D1 Hayes Lab
supIPDF D5-1 Hayes Lab
supIPDF D5-2 Hayes Lab](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-24-320.jpg)
![Hunter 25
Table 4: Primers used for plaque PCR
Table 5: In vivo complementation data for i434Dam123 plating on fusion host strains at
different temperatures
i434Dam123 25ºC 30ºC 37ºC 41ºC
594[p613] 0 2.3*104 3*109 2.5*109
TC600[p613] TNTC TNTC 2.1*109 2*109
594[p613*] 0 4.9*103 0 0
TC600[p613*] TCL TCL 1.9*109 2.4*109
594[p614] 6*102 2.3*103 1.1*109 1.1*109
TC600[p614] TCL TCL 2*109 2.5*109
594[p614*] 0 7.9*103 0 0
TC600[p614*] TCL TCL 1.7*109 2*109
594[p617] 0 4.9*103 0 0
TC600[p617] TCL TCL 1.5*109 1.4*109
594[p623] 0 5.5*103 0 0
TC600[p623] TCL TCL 1.4*109 1.8*109
594[p625] 1.7*103 5.8*103 0 0
TC600[p625] TCL TCL 2.2*109 2*109
594[p628] 1.6*103 4.3*103 0 0
TC600[p628] TCL TCL 1.3*109 1.1*109
Primers Direction Primer sequence
Primer #1 LNu3forD (at 5511bp for λ) Leftward tcaactgtgaggaggctcac
Primer #4 RendEforD (at 6327bp for λ) Rightward RC acggataacctcaccggaaaca
Primer #2 LDforE (at 5991bp for λ) Leftward gttatgaggatgtgctctgg
Primer #5 RfiforE (at 7254bp for λ) Rightward RC ttcagttgttcacccagcg
Primer #3 LNendEforE (at 6230bp for λ) Leftward gagagctatcccttcacca](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-25-320.jpg)
![Hunter 26
Table 6: In vivo complementation data for i434(1812)p22 plating on fusion host strains at
different temperatures
i434(1812)p22 25ºC 30ºC 37ºC 41ºC
594[p613] TCL TCL 1.5*109 1.5*109
TC600[p613] TCL TCL 1.2*109 1.1*109
594[p613*] TCL TCL 1.2*109 1.4*109
TC600[p613*] TCL TCL 1.4*109 1.4*109
594[p614] TCL TCL 7.3*108 4.9*108
TC600[p614] TCL TCL 1.2*109 8.7*108
594[p614*] TCL TCL 1.2*109 1.4*109
TC600[p614*] TCL TCL 1*109 1.2*109
594[p617] TCL TCL 9.7*108 9.5*108
TC600[p617] TCL TCL 8.3*108 9.2*108
594[p623] TCL TCL 1.4*109 9.1*108
TC600[p623] TCL TCL 1.2*109 1*109
594[p625] TCL TCL 9.3*108 1*109
TC600[p625] TCL TCL 1*109 1.2*109
594[p628] TCL TCL 8.3*108 8.8*108
TC600[p628] TCL TCL 1.2*109 1.1*109
Table 7: i434Dam123 plaques picked from 594 onto 594 and 594[p617] to select for IPDF
mutants
+3= large plaques (>5mm)
+2= medium plaques (3-5m)
+1= small plaques (1-2mm)
(-) = no plaque
38ºC
594 594[p617]
1 2 3 4 5 1 2 3 4 5
A +3 +3 +3 +3 +3 - +3 +3 +3 +1
B +3 +3 +3 +3 +3 +3 +3 +3 - +3
C +3 +3 +3 +3 +3 +3 +3 +3 +3 +3
D +3 +3 +3 +3 +3 +1 +3 +3 +3 -](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-26-320.jpg)
![Hunter 27
Table 8: Plaque picking assay for the isolation of IPDF mutants
Table 9: Titres for the primary phage lysates of plaques picked from the 594 strip plates of
the mutants isolated from the “t” column of Set 2
38ºC IPDF A1-t IPDF A5-t IPDF B4-t IPDF D1-t IPDF D5-t
594
10-7 986 683 881 174 509
10-8 116 67 69 30 57
Titre 1*1010 6.8*109 7.9*109 2.4*109 5.4*109
594[p617]
10-2 7 0 43 TCL1. 7
10-3 0 0 6 TCL 1
10-4 0 1 0 TCL 0
Titre 7*102 <102 5.2*102 >104 7*102
Reversion Frequency 7*10-8 1.5*10-8 6.6*10-7 Nd2. 1.3*10-7
1. TCL= Total Cell Lysis
2. ND= Not determined
38ºC 594594 594594[p617]
Set 1
O N M L K O N M L K
A1 A1
B4
B4
D5
D5
A5
A5
D1 D1
T S R Q P T S R Q P
Set 2
A1 A1
B4 B4
D5 D5
A5 A5
D1 D1](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-27-320.jpg)
![Hunter 28
Table 10: Plaques from IPDF lysates plated on 594[p617] and picked onto 594[p617] for
isolation of supIPDF mutants
594[p617] A1-t A5-t D1-t B4-t D5-t
38ºC A1-1 A1-2 A5-1 A5-2 D1-1 B4-1 B4-2 B4-3 D5-1 D5-2
1 (-) (-) (-) (-) +3 +3 (-) (-) +3 +3
2 (-) (-) (-) (-) +3 +3 +3 (-) +3 +3
3 (-) (-) (-) (-) +3 +3 +3 (-) +3 +3
4 (-) (-) (-) (-) +2 +1 +3 (-) +3 +3
5 (-) (-) (-) (-) +3 (-) +2 (-) +3 +3
Table 111.: Plaques exhibiting supIPDF phenotype picked from 594[p617] plate onto 594
594
38ºC B4-1 B4-2 D1-1 D5-1 D5-2
1 +3 clear/turbid -- +3 +3 +3 clear/turbid
2 +3 clear +3 +3 +3 +3
3 +3 +3 +3 +3 clear/turbid +3
4 +3 +3 +3 +3 +3 clear/turbid
5 -- +3 +3 +3 +3 clear/turbid
1. Unless specified otherwise, plaques on 594 were turbid.
Table 12: Plaques exhibiting supIPDF phenotype picked from 594[p617] plate onto
594[p617]
594[p617]
38ºC B4-1 B4-2 D1-1 D5-1 D5-2
1 +3 -- +3 +3 +3
2 +3 +3 +3 +3 +3
3 +3 +3 +3 +3 +3
4 +3 +3 +3 +3 +3
5 -- +3 +3 +3 +3](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-28-320.jpg)
![Hunter 29
Table 13: Titres for supIPDF mutants isolated from 594 strip plates and plated onto 594
and 594[p617]
38ºC supIPDF B4-2-2 supIPDF D1-1-1 supIPDF D5-1-1 supIPDF D5-2-2
594
10-7 455 580 251 255
10-8 56 60 24 28
Titre 5*109 5.9*109 2.5*109 2.7*109
594[p617]
10-7 132 343 92 159
10-8 22 29 15 26
Titre 1.8*109 3.2*109 1.2*109 2.1*109
Figure 1: (A) Visual map of a 816bp fragment overlapping from part of Nu3 to part of E in λ
along with positions for Primers 2, 3, and 4 for sequencing of D in IPDF and supIPDF mutants.
Coloured segments below the sequence illustrate relative positions of Nu3, D, an intergenic
region, and E. (B) Visual map of a 1264bp fragment overlapping from part of D to part of E in λ
along with positions for primers 2, 3, 4, and 5 for sequencing of E in IPDF and supIPDF mutants.
Coloured segments below the sequence illustrate the relative positions of D, an intergenic region,
and E.
A.
B.](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-29-320.jpg)
![Hunter 30
Table 14: Titres of IPDF and supIPDF phage strains at 38ºC
Phage Lysate Plating on 594 (PFU/ml) Plating on 594[p617] (PFU/ml)
IPDF A1 1*1010 7*102
IPDF A5 6.8*109 <102
IPDF B4 7.9*109 5.2*103
IPDF D5 5.4*109 7*102
IPDF D1 2.4*109 >104
SupIPDF B4 5*109 1.8*109
SupIPDF D1 5.9*109 3.2*109
supIPDF D5-1 2.5*109 1.2*109
SupIPDF D5-2 2.7*109 2.1*109
Table 15: Titres of #1027 i434Dam123 plated on different host strains to detect vir
contaminants at 30ºC
#1027 i434Dam123
30ºC TC600 594 TC600[λpapa] TC600[i434T]
10-2 -- 34 TCL1 2
10-3 -- 4 TCL 0
10-4 -- 0 TCL 0
Titre -- 3.7*103 ND 2*102
10-6 TNTC -- TNTC2 0
10-7 193 -- 273 0
10-8 23 -- 22 0
Titre 2.1*109 -- 2.5*109 ND3
1. TCL is Total Cell Lysis
2. TNTC is Too Numerous to Count
3. ND is Not Determined](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-30-320.jpg)
![Hunter 31
Table 16: Titre of MMS 179 #1027b i434Dam123 plated on different host strains to detect
virλ contaminants
MMS 179 #1027b i434Dam123
30ºC TC600 594 TC600[λpapa] TC600[i434T]
10-2 -- 186 -- 0
10-3 -- 154 -- 0
10-4 -- 0 -- 0
Titre -- 1.9*103 -- <102
10-6 134 -- 243 --
10-7 24 -- 29 --
10-8 3 -- 2 --
Titre 2.2*108 -- 2.4*108 --
Table 17: Sequencing results for E of IPDF mutants
IPDF A1 IPDF A5 IPDF B4 IPDF D1 IPDF D5
Primer #2
LDforE
145-810bp 285-799bp 140-880bp 361-900bp 120-840bp
6135-6800bp
good for E
6275-6789bp
good for E
6130-6870bp
GA at
6860bp
6351-6890bp G
is deleted at
873bp
6110-6830bp
GA at 812bp
CT at 821bp
Primer #3
LNendEforE
513-1170bp 291-1032bp 361-960bp 301-1140bp 301-1140bp
6503-7142bp
good for E
CG at
6968bp
6281-7022bp
good for E
6351-6950bp
GC at
6968bp
6291-7130bp A
insertion at
1032bp
6291-7130bp
CT
Primer #5
RfiforE
508-1170bp 361-1231bp 421-1200bp 301-1200bp 481-1200bp
6499-7157bp
good for E
6352-7218bp
good for E
6410-7190bp
GA at
6511bp
6291-7190bp C
insertion at
6313bp
CG at 412bp
6471-7190bp
good for E
Conclusion
IPDF A1-t is
wildtype for E
IPDF A5-t is
wildtype for E
IPDF B4-t is
wildtype for E
IPDF D1-t is
wildtype for E
IPDF D5-t is
wildtype for E](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-31-320.jpg)

![Hunter 33
Table 20: Plating IPDF D1 and supIPDF lysates on 594[p613] at 30ºC and 40ºC to assess
effects of cI repression on plating efficiency
594[p613] 30ºC 40ºC
Fold increase in plating
(Titre at 40ºC/Titre at 30ºC) Conclusion
SupIPDF D5-2
10-7 184 229
1.14X
supIPDF D5-2
plating is
independent of
exogenously
expressed gpD
10-8 24 26
Titre 2.1*109 2.4*109
supIPDF B4 supIPDF B4
plating is
independent of
exogenously
expressed gpD
10-7 279 427
1.6X
10-8 31 52
Titre 2.9*109 4.7*109
SupIPDF D5-1 supIPDF D5-1
plating is
independent of
exogenously
expressed gpD
10-7 184 329
1.89X
10-8 18 36
Titre 1.8*109 3.4*109
SupIPDF D1 supIPDF D1
plating is
independent of
exogenously
expressed gpD
10-7 450 628
1.44X
10-8 42 61
Titre 4.3*109 6.2*109
IPDF D1 IPDF D1
plating is
independent of
exogenously
expressed gpD
10-7 197 368
1.4X
10-8 35 39
Titre 2.7*109 3.8*109](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-33-320.jpg)
![Hunter 34
Table 21: Plating IPDF D1 and supIPDF lysates on 594[p613*] at 30ºC and 40ºC to assess
effects of cI repression on plating efficiency
594[p613*] 30ºC 40ºC
Fold increase in plating
(Titre at 40ºC/Titre at 30ºC) Conclusion
SupIPDF D5-2
10-7 131 266
1.28X
SupIPDF D5-2
plating is
independent of
exogenously
expressed
67bpΔgpD
10-8 30 27
Titre 2.1*109 2.7*109
supIPDF B4 SupIPDF B4
plating is
independent of
exogenously
expressed
67bpΔgpD
10-7 218 547
2X
10-8 32 54
Titre 2.7*109 5.4*109
SupIPDF D5-1 SupIPDF D5-1
plating is
independent of
exogenously
expressed
67bpΔgpD
10-7 111 329
3X
10-8 14 47
Titre 1.3*109 4*109
SupIPDF D1 SupIPDF D1
plating is
independent of
exogenously
expressed
67bpΔgpD
10-7 198 592
2.56X
10-8 30 69
Titre 2.5*109 6.4*109
IPDF D1 IPDF D1
plating is
independent of
exogenously
expressed
67bpΔgpD
10-7 84 401
3.17X
10-8 16 35
Titre 1.2*109 3.8*109](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-34-320.jpg)
![Hunter 35
Table 22: Plating IPDF D1 and supIPDF lysates on 594[p626] at 30ºC and 40ºC to assess
effects of cI repression on plating efficiency
594[p626] 30ºC 40ºC
Fold increase in plating
(Titre at 40ºC/Titre at
30ºC)
Conclusions
SupIPDF D5-2
10-7 147 213
1.59X
supIPDF D5-2 plating is
independent of an exogenously
expressed gpD-fusion
10-8 14 24
Titre 1.45*109 2.3*109
SupIPDF B4
supIPDF B4 plating is
independent of an exogenously
expressed gpD-fusion
10-7 293 300
1.39X10-8 28 43
Titre 2.8*109 3.9*109
SupIPDF D5-11
supIPDF D5-1 plating is
independent of an exogenously
expressed gpD-fusion
10-7 211 254
1.3X10-8 28 33
Titre 2.1*109 2.7*109
SupIPDF D1
supIPDF D5-2 plating is
independent of an exogenously
expressed gpD-fusion
10-7 277 447
1.47X10-8 43 61
Titre 3.2*109 4.7*109
IPDF D1
IPDF D1 plating is independent of
an exogenously expressed gpD-
fusion
10-7 228 292
1.25X10-8 32 31
Titre 2.4*109 3*109
1. Plating for supIPDF D5-1 was carried out separately and on a different day than the other
supIPDF lysates. Procedure and cultures used were identical.](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-35-320.jpg)
![Hunter 36
Table 23: Secondary lysates (Output Phage) plated on 594 and TC600 to calculate marker
rescue frequency
Table 24: Primary lysates (Input Phage) plated on 594 and TC600 to calculate reversion
frequency
30ºC i434Dam123 Reversion frequency from input phage
594
1.46*10-6
10-1 293turbid plaques
10-2 35
10-3 5
Titre 3.2*103
TC600
10-6 Approx. 2420
10-7 205
10-8 18
Titre 2.2*109
Table 25: Percent increase in reversion frequency due to marker rescue
Marker rescue
frequency from
output phage
Reversion
frequency from
input phage
Percent change in
frequency (%)
(Marker rescue
frequency/Reversion
frequency)
Secondary lysate
i434Dam123 lysate
grown on 594[p613]
4*10-6 1.46*10-6 273%
30ºC
Secondary lysate i434Dam123
lysate grown on 594[p613]
Secondary lysate i434Dam123
grown on 594[p613*]
594
10-1 1 0
10-2 1 0
10-3 0 0
Titre 10 <10
TC600
10-4 268 226
10-5 24 18
10-6 1 1
Titre 2.5*106 2*106
Marker rescue frequency
from output phage
4*10-6 <5*10-6](https://image.slidesharecdn.com/4d3dd7de-89b8-4138-9da3-84d2243aa7ff-151207164441-lva1-app6892/85/Honours-Report-Draft-COMPLETE-36-320.jpg)
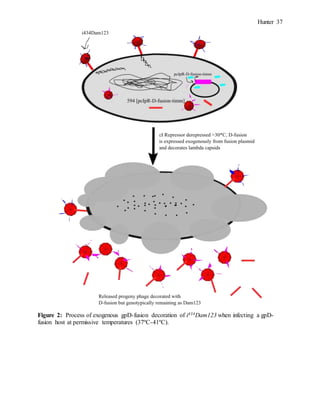




This document describes experiments aimed at isolating bacteriophage mutants with altered tolerance for plating when the gpD capsid protein is fused to foreign proteins. The author isolated "IPDF" mutants of phage i434Dam123 that could not plate when gpD-fusions were expressed from a plasmid. These IPDF mutants were then used to select "supIPDF" mutants that regained the ability to plate equally in the presence or absence of gpD-fusions. Sequencing showed the mutations responsible for the IPDF and supIPDF phenotypes were located outside of the D and E genes. This suggests there are extragenic mutations that can enhance or suppress the toxicity of gpD-fusions towards viable phage















































